
线粒体ROS与心房颤动*
2017-10-20 05:28:40郑安财李菊香
中国病理生理杂志 2017年10期
郑安财, 李菊香
(南昌大学第二附属医院心内科, 江西 南昌 330006)
线粒体ROS与心房颤动*
郑安财, 李菊香△
(南昌大学第二附属医院心内科, 江西 南昌 330006)
心房颤动(atrial fibrillation,AF)是临床上最为常见的快速性心律失常之一。在过去的几十年中,AF的患病率随年龄的增长而稳步增加。AF可以引起心房内血栓、脑栓塞等严重并发症,是患者死亡的重要原因。最近有报道称,线粒体氧化应激参与AF的心电重构过程和氧化还原反应的调控[1]。本文将探讨线粒体氧化应激的形成及其通过活性氧簇(reactive oxygen species,ROS)影响重要的离子通道、促进心房电生理与结构重构,从而最终导致房颤发生的机制。
1线粒体与ROS

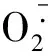
2ROS与心房颤动
近年来,在AF的基础及临床研究中发现,AF可引起心房动作电位(action potential duration,APD)、有效不应期(effective refractory period,ERP)、有效不应期频率适应性(effective refractory period-rate adaptation,ERP-RA)和心房传导速度的变化,即心房电重构,后者反过来又促进AF的发作和维持。线粒体氧化应激可促进AF时的心房重构,但其分子机制目前尚未明确。正常情况下,线粒体产生的ATP被细胞膜、内质网离子通道及转运蛋白所利用,以保证心肌细胞电活动所必需的能量。线粒体功能障碍则减少心肌细胞离子通道和转运体的能量供应,从而导致心律紊乱。最新研究表明,过多的线粒体ROS可通过胱氨酸转录后的氧化还原修饰(如蛋白质谷胱甘肽化)或酪氨酸残基的硝化反应,直接影响各种离子通道和转运蛋白,减弱心肌兴奋性[10],从而导致心房APD延长,引起早期后除极(early afterdepolarizations,EADs)和延迟后除极(delayed afterdepolarizations,DADs),三者共同促进异位活动及折返心律的形成,而异位活动及折返已被确切地认为是AF的主要电生理机制。
3ROS调节离子通道及转运蛋白
已证明对氧化还原敏感的Na+、Ca2+及K+离子调控蛋白能够影响心肌兴奋-收缩耦联的过程,因此,调控蛋白的异常可影响心脏功能并导致心律失常,特别是AF。在心肌细胞的动作电位形成中,电压门控Na+通道引起膜去极化,激活L型Ca2+通道(L-type calcium channel,LTCCs)产生跨膜Ca2+内流(ICa,L),ICa,L通过触发心脏肌浆网Ca2+释放通道兰尼碱受体2(ryanodine receptor 2,RyR2),使肌浆网(sarcoplasmic reticulum,SR)储存的大量Ca2+释放,成为Ca2+诱导的Ca2+释放。Ca2+与肌钙蛋白C结合,导致肌球蛋白ATP酶激活,形成收缩。当Ca2+通过Na+-Ca2+交换和细胞膜Ca2+-ATP酶自细胞内向外转运以及肌浆/内质网Ca2+-ATP酶2a(sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2a,SERCA2a)再次储存于肌浆网后,心房肌细胞舒张。这些离子调控蛋白主要受磷酸化作用的调节,因此,氧化还原敏感性蛋白激酶,如钙离子/钙调素依赖性蛋白激酶II(Ca2+/calmodulin-dependent protein kinase II,CaMKII)、蛋白激酶C(protein kinase C,PKC)、蛋白激酶A(protein kinase A,PKA)等的激活将极大地影响心肌细胞内的离子平衡和功能。
3.1ROS与Ca2+调控 Ca2+调控异常是心房电重构的主要表现之一。胞内Ca2+水平的变化有助于AF的诱发和维持,且是AF从阵发性转为持续性的必不可少的机制[11]。细胞内Ca2+浓度受到各种Ca2+调控蛋白的调节,包括L型Ca2+通道、肌浆网Ca2+-ATP酶、肌浆网Ca2+释放通道、Na+-Ca2+交换体(Na+/Ca2+exchanger,NCX)和多种信号分子如CaMKII、PKC和PKA。这些Ca2+调控蛋白的蛋氨酸残基及硫醇易受ROS或还原剂的直接调节。研究发现,应用H2O2或增加线粒体ROS能增大ICa,L,溶血卵磷脂(lysophosphatidylcholine,LPC)诱导增加的线粒体ROS能氧化低密度脂蛋白(low-density lipoprotein,LDL),氧化的LDL能提高ICa,L[12]。CaMKII也能被ROS激活,激活的CaMKII能磷酸化Cav1.2通道亚基,并增加其开放几率,从而增大ICa,L。最近研究报道,胞质内NADH/NAD+氧化还原反应对在调节NCX活性中扮演重要角色,钠钙交换电流(so-dium-calcium exchange current,INCX)可被NADH抑制,而且这种抑制状态可被过氧化氢酶解除[13]。ROS亦能激活反向型NCX,增加Ca2+内流,产生短暂INCX,导致复极4期膜去极化,即DADs;此外,大量ROS造成的线粒体ATP合成障碍,使SERCA2a活性下降,从而导致SR的Ca2+储存减少[14]。RyR2功能紊乱引起的舒张期肌浆网Ca2+渗漏是导致AF时钙调控紊乱的一个重要因素。在小鼠的AF模型以及持续性AF患者中,RyR2的开放几率显著增加,过量的ROS增加RyR2通道的开放几率,从而增加Ca2+从SR上渗漏,形成钙火花[15]。Shan等[16]发现RyR2被氧化后,能够增强PKA介导的稳钙蛋白FKBP12.6从RyR2复合体上解离,使RyR2结构不稳定,促进Ca2+渗漏。ROS激活的CaMKII也能磷酸化RyR2的Ser2814位点,增加SR的Ca2+渗漏,促进胞质Ca2+超载及DADs[17]。另外,Ca2+过载不仅造成线粒体ATP合成障碍,还能影响线粒体膜电位,促进线粒体内NO和ROS产生,进一步诱导ROS的堆积[18]。因此,在ROS诱导Ca2+过载和Ca2+过载诱导ROS堆积间,将形成一个恶性循环。这个正反馈回路将压倒体内对ROS和Ca2+的清除能力,造成细胞损伤和折返电位,促进房颤的发生。总之,胞内过量ROS对Ca2+调控蛋白的影响将使细胞内Ca2+过载及SR的Ca2+储存减少,导致DADs和收缩功能障碍,促进AF的发生。

3.3ROS与Na+调控 ROS已被证实可通过多种途径调节Na+通道功能。转录水平上,ROS通过影响mRNA的转录,减少电压门控钠离子通道Nav1.5的表达;蛋白水平上,Nav1.5通道的甲硫氨酸残基具有氧化敏感性,被ROS氧化后,将显著减缓Nav1.5的失活,增强晚钠电流,最终导致传导阻滞,诱发折返性心律失常[21]。线粒体ROS增加一方面可在不改变细胞膜Nav1.5通道表达的前提下,通过调节Nav1.5通道的传导性,降低峰钠电流,导致传导阻滞[22];另一方面可激活PKC,后者可能通过影响Nav1.5通道的磷酸化,降低峰钠电流[23]。ROS也可通过氧化激活CaMKII增强晚钠电流,从而导致APD延迟,诱发EADs[24]。
4针对线粒体的抗氧化治疗防治心房颤动

5小结和展望
线粒体ROS在房颤的发生、发展中发挥重要的作用,当机体处于病理状态时,线粒体功能受损导致ROS大量产生,进而通过氧化还原反应激活信号分子,改变心肌细胞内多种离子通道和转运蛋白功能,通过多种机制促进AF的发生发展。因此,对线粒体ROS及功能进行深入研究以创制出线粒体靶向抗氧化剂,可从源头上减少ROS的产生,保护线粒体功能,从而减轻线粒体活性氧对心血管的损伤,将使更多的房颤患者获益。尽管到目前为止,抗氧化治疗的效果不理想,但针对线粒体的抗氧化治疗仍是预防和治疗AF的一个合理方向。对线粒体ROS进行深入研究并开展大量临床实验,将会为AF的防治提供新的思路和途径。
[1] Yang KC, Dudley SC Jr. Oxidative stress and atrial fibrillation: finding a missing piece to the puzzle[J]. Circulation, 2013, 128(16):1724-1726.
[2] Bleier L, Dröse S. Superoxide generation by complex III: from mechanistic rationales to functional consequences[J]. Biochim Biophys Acta, 2013, 1827(11-12):1320-1331.
[3] Krivoruchko A, Storey KB. Forever young: mechanisms of natural anoxia tolerance and potential links to longevity[J]. Oxid Med Cell Longev, 2010, 3(3):186-198.
[4] Panov A, Dikalov S, Shalbuyeva N, et al. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice[J]. Am J Physiol Cell Physiol, 2007, 292(2):C708-C718.
[5] Nazarewicz RR, Dikalova AE, Bikineyeva A, et al. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress[J]. Am J Physiol Heart Circ Physiol, 2013, 305(8):H1131-H1140.
[6] Fang H, Chen M, Ding Y, et al. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals[J]. Cell Res, 2011, 21(9):1295-1304.
[7] Xie C, Kauffman J, Akar FG. Functional crosstalk between the mitochondrial PTP and KATPchannels determine arrhythmic vulnerability to oxidative stress[J]. Front Physiol, 2014,5:264.
[8] Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release[J]. Physiol Rev, 2014, 94(3):909-950.
[9] Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination[J]. J Mol Cell Cardiol, 2014, 73:26-33.
[10]Aggarwal NT, Makielski JC. Redox control of cardiac excitability[J]. Antioxid Redox Signal, 2013, 18(4):432-468.
[11]Schotten U, Verheule S, Kirchhof P, et al. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal[J]. Physiol Rev, 2011, 91(1):265-325.
[12]Fearon IM. OxLDL enhances L-type Ca2+currents via lysophosphatidylcholine-induced mitochondrial reactive oxygen species (ROS) production[J]. Cardiovasc Res, 2006, 69(4):855-864.
[13]Liu T, O′Rourke B. Regulation of the Na+/Ca2+exchanger by pyridine nucleotide redox potential in ventricular myocytes[J]. J Biol Chem, 2013, 288(44):31984-31992.
[14]Yang KC, Bonini MG, Dudley SC Jr. Mitochondria and arrhythmias[J]. Free Radic Biol Med, 2014, 71:351-361.
[15]Xie W, Santulli G, Reiken SR, et al. Mitochondrial oxidative stress promotes atrial fibrillation[J]. Sci Rep, 2015, 5:11427.
[16]Shan J, Betzenhauser MJ, Kushnir A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice[J]. J Clin Invest, 2010, 120(12):4375-4387.
[17]Li N, Wang T, Wang W, et al. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice[J]. Circ Res, 2012, 110(3):465-470.
[18]Dedkova EN, Blatter LA. Characteristics and function of cardiac mitochondrial nitric oxide synthase[J]. J Physiol, 2009, 587(Pt 4):851-872.
[19]Asada K, Kurokawa J, Furukawa T. Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel[J]. J Biol Chem, 2009, 284(9):6014-6020.
[20]Svoboda LK, Reddie KG, Zhang L, et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5[J]. Circ Res, 2012, 111(7):842-853.
[21]Kassmann M, Hansel A, Leipold E, et al. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation[J]. Pflugers Arch, 2008, 456(6):1085-1095.
[22]Liu M, Gu L, Sulkin MS, et al. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy[J]. J Mol Cell Cardiol, 2013, 54:25-34.
[23]Liu M, Liu H, Dudley SC Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel[J]. Circ Res, 2010, 107(8):967-974.
[24]Wagner S, Ruff HM, Weber SL, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for lateINaaugmentation leading to cellular Na and Ca overload[J]. Circ Res,2011,108(5):555-565.
[25]Smith RA, Hartley RC, Cochemé HM, et al. Mitochondrial pharmacology[J]. Trends Pharmacol Sci, 2012, 33(6):341-352.
[26]Prime TA, Blaikie FH, Evans C, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury[J]. Proc Natl Acad Sci U S A, 2009, 106(26):10764-10769.
(责任编辑:林白霜, 余小慧)
Mitochondrialreactiveoxygenspeciesandatrialfibrillation
ZHENG An-cai, LI Ju-xiang
(DepartmentofCardiology,TheSecondAffiliatedHospital,NanchangUniversity,Nanchang330006,China.E-mail:ljx912@126.com)
Atrial fibrillation (AF) is the most common arrhythmia in clinical practice. Mitochondrial oxidative stress is supposed to contribute to development, progression and self-perpetuation of AF. Reactive oxygen species (ROS) is the major molecule mediating mitochondrial oxidative stress damage. ROS can alter the redox status of various molecular targets, which quite specifically leads to functional alterations of ion channel activity or activation of a variety of redox sensitive signal transduction pathways. Eventually, it leads to atrial electrical remodeling and promotes the development of AF. Therefore, mitochondrial oxidative stress pathways may be a new target for the therapy of atrial fibrillation.
线粒体; 活性氧簇; 离子通道; 心房颤动
Mitochondrial; Reactive oxygen species; Ion channel; Atrial fibrillation
R541; R363
A
10.3969/j.issn.1000- 4718.2017.10.031
1000- 4718(2017)10- 1917- 04
2017- 05- 04
2017- 07- 18
江西省自然科学基金重大项目(No. 20152ACB20025);江西省赣鄱555人才计划项目
△通讯作者 Tel: 0791-88060095; E-mail: ljx912@126.com
杂志网址: http://www.cjpp.net
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:17:14
中国医药导报(2021年33期)2021-12-26 01:58:24
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:36
科学(2020年2期)2020-08-24 07:57:00
山东医药(2018年36期)2018-11-02 01:26:26
中国循环杂志(2015年10期)2015-12-24 03:29:57
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:17
医学研究杂志(2015年3期)2015-06-10 06:41:52
中国病理生理杂志(2015年10期)2015-01-26 04:36:31
西南军医(2014年6期)2014-01-22 06:57:43
