
氯沙坦的合成工艺改进
2017-10-13 03:00:27臧永军韩邦兴
合成化学 2017年10期
臧永军, 刘 东, 韩邦兴
(1.皖西学院 a. 生物与制药工程学院; b. 中药研究与开发工程技术研究中心,安徽 六安 237012)
·制药技术·
氯沙坦的合成工艺改进
臧永军1a, 刘 东1a, 韩邦兴
(1.皖西学院 a. 生物与制药工程学院; b. 中药研究与开发工程技术研究中心,安徽 六安 237012)
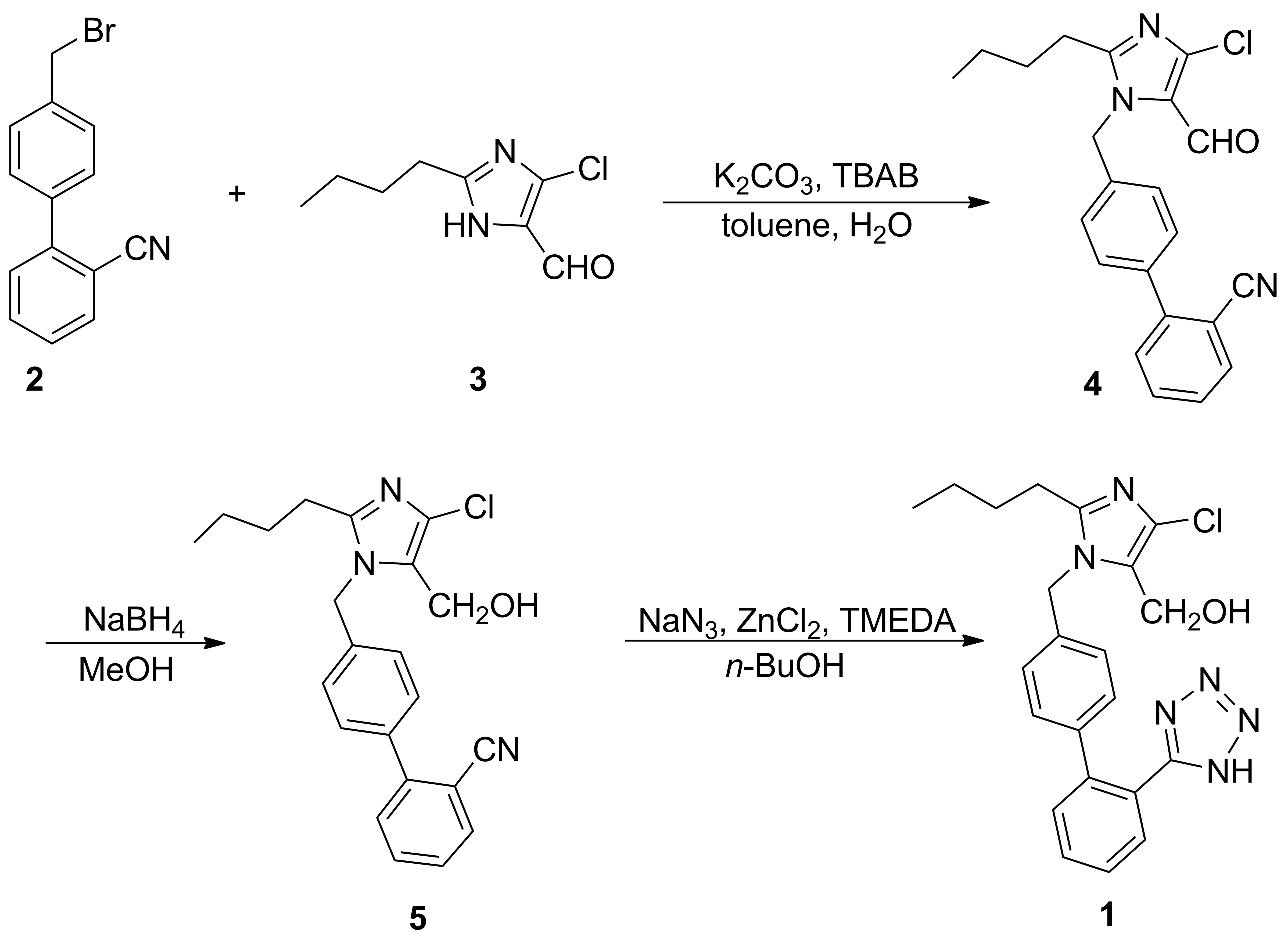
以2-氰基-4′-溴甲基联苯和2-丁基-4-氯-5-甲酰基-咪唑为原料,依次经N-烷基化、羟基化和四唑化反应合成了氯沙坦,总收率73.9%,其结构经1H NMR和MS(ESI)确证。
2-氰基-4′-溴甲基联苯; 氯沙坦; 四氮唑化反应; 药物合成; 工艺改进
Abstract: Losartan, with an overall yield of about 73.9%, was synthesized from 2-n-butyl-4-ehloro-5-imidazoleearboxaldehyde and 2-cyano-4′-bromomethylbiphenylviathe reaction ofN-alkyltion, hydroxylation and tetrazolation in sequence. The structure was confirmed by1H NMR and MS(ESI).
Keywords: 2-cyano-4′-bromomethylbiphenyl; losartan; tetrazolation; drug synthesis; process improvement
氯沙坦(1)钾盐是首种上市的沙坦类降压药物。1主要通过选择性和竞争性结合AngII受体亚型AT1,从而阻断AngII与AT1的结合,最终发挥降压、保护器官的作用[1]。氯沙坦钾由美国Bristol公司研发,首先于1994年在瑞典上市,1995年获FDA批准后相继在多个国家上市。药理和临床试验结果均表明:氯沙坦钾具有服药方便、作用广泛、降压作用显著、对肾功能影响小等优点,是一种安全的高效降压药物[2]。
目前,1的合成方法主要有以下3种:(1)2-氰基-4′-溴甲基联苯(2)与叠氮钠发生四唑化反应,用三苯甲基氯对四唑环的N—H进行保护,经溴代制得中间体5-(4-溴甲基联苯-2-基)-1-三苯甲基-1H-四唑,然后与2-丁基-4-氯-5-甲酰基-咪唑(3)发生N-烷基化反应,再经羟基化和脱保护反应合成1[3-6]。 (2)2-丁基-4-氯-5-羟甲基咪唑与对溴苄溴反应制得[3-(4-溴-1-苄基)-2-丁基-5-氯-3H-咪唑-4-基]甲醛,再与2-(1H-四氮唑-5-基)苯硼酸反应生成三苯基氯沙坦,然后再经羟基化和脱保护基反应制得1[7-9]。(3)以2和3为原料,依次经N-烷基化、羟基化和四唑化反应制得1[10-13]。
路线(1)和(2)均是先进行四唑化反应,需经过四唑环的保护与脱保护过程,成本较高,步骤繁杂, 收率偏低。路线(3)以廉价易得的中间体为原料,路线较短,收率较高,已实现工业化。但在
Scheme1
四唑化过程中需使用价格昂贵且毒性较大的有机锡卤化物或三乙胺盐酸盐作催化剂。此外,还存在叠氮化钠用量较大(4 eq.),产品色泽差,需经多次脱色等缺点。
本文在路线(3)的基础上,参考文献[10-11]方法,以2和3为原料,依次经N-烷基化、羟基化和四唑化反应合成了1(Scheme 1),总收率73.9%,其结构经1H NMR和MS(ESI)确证
1 实验部分
1.1 仪器与试剂
B-540型熔点仪(温度未校正);Varian-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Trace DSQ FINNIGSN型质谱仪。
2,3和叠氮化钠,浙江华海药业,用前未经处理;其余试剂均为分析纯。
1.2 合成
(1) 2-丁基-4-氯-5-羟甲基-1-(2′-氰基-联苯基-4-)甲基咪唑(5)的合成
在三口烧瓶中依次加入275.0 g(0.265 mol),346.5 g(0.250 mol),碳酸钾48.3 g(0.350 mol),四丁基溴化铵(TBAB)4.00 g,甲苯240 mL和水80 mL,搅拌下于35 ℃反应10 h(TLC跟踪,展开剂:石油醚/乙酸乙酯=3/1,V/V)。分液,除去水层,有机层依次用5%NaOH溶液(120 mL)和水(120 mL)洗涤,加入甲醇80 mL和硼氢化钠3.80 g(0.100 mol),搅拌下于40 ℃反应1 h。加水320 mL,于60 ℃反应1 h;冰浴冷却1 h,析出产品,过滤,滤饼水洗后,经干燥得白色粉末581.5 g,收率87.0%, m.p.159.2~160.4 ℃(159.0~160.0 ℃[7]);1H NMRδ: 7.73(d,J=8.0 Hz, 1H), 7.63(t,J=8.0 Hz, 1H), 7.50(d,J=8.0 Hz, 2H), 7.47~7.41(m, 2H), 7.11(d,J=8.0 Hz, 2H), 5.36(s, 2H), 4.49(s, 2H), 3.80(s, 1H), 2.56(t,J=4.0 Hz, 2H), 1.67~1.61(m, 2H), 1.37~1.28(m, 2H), 0.86(t,J=6.0 Hz, 3H); MS(ESI)m/z: 380.6{[M+H]+}。
(2)1的合成
在三口瓶中依次加入NaN319.5 g(0.300 mol), ZnCl229.7 g(0.220 mol)和正丁醇90 mL,于85 ℃缓慢滴加N,N,N′,N′-四甲基乙二胺(TMEDA)25.5 g(0.220 mol)的正丁醇(70 mL)溶液,滴毕(30 min),搅拌下反应1 h。加入570.0 g(0.184 mol),回流(110 ℃)反应40 h(TLC跟踪,展开剂:二氯甲烷/甲醇=10/1,V/V)。冷却至室温,加入15%NaOH溶液150 mL,搅拌1 h;滤除Zn(OH)2,滤液分液,除去水层,有机相用饱和食盐水(2×200 mL)洗涤,用6 mol·L-1盐酸调至pH 3.5,于室温剧烈搅拌1 h。抽滤,滤饼依次用乙酸乙酯(2×40 mL)和水(2×40 mL)洗涤,于45 ℃真空干燥得白色固体166.2 g,收率85.0%, m.p.184.2~186.5 ℃(184.3~186.0 ℃[7]);1H NMRδ: 16.0(br, 1H), 7.60~7.50(m, 2H), 7.40(d,J=9.0 Hz, 2H), 7.14~7.03(dd,J=8.0 Hz, 8.0 Hz, 4H), 5.23(s, 2H), 4.49(s, 2H), 2.61(t,J=7.0 Hz, 2H), 1.68~1.62(m, 2H), 1.38~1.32(m, 2H), 0.88(t,J=7.0 Hz, 3H); MS(ESI)m/z: 421.1{[M+H]+}。
合成.5时,文献[10-12]方法均是先将N-烷基化产物分离纯化后再进行羟基化。我们采用 “一锅烩”的方法,即将N-烷基化产物直接加入下一步羟基化反应。合成1时,以TMEDA为配体,与NaN3和ZnCl2先形成配合物,再与5进行四唑化反应,总收率73.9%。
为提高5的收率,我们对5的合成条件进行了优化。表1为溶剂对5收率的影响。由表1可见,以NMP或DMF为溶剂,反应时间短(3 h),收率较高,但工业生产中DMF与NMP不易回收,对环境污染大,不宜选用。以EtOAc/H2O为溶剂,反应时间长,收率较低。以toluene/H2O作溶剂(V/V=3/1),收率较高(87%)。因此,选择toluene/H2O(V/V=3/1)为反应溶剂。
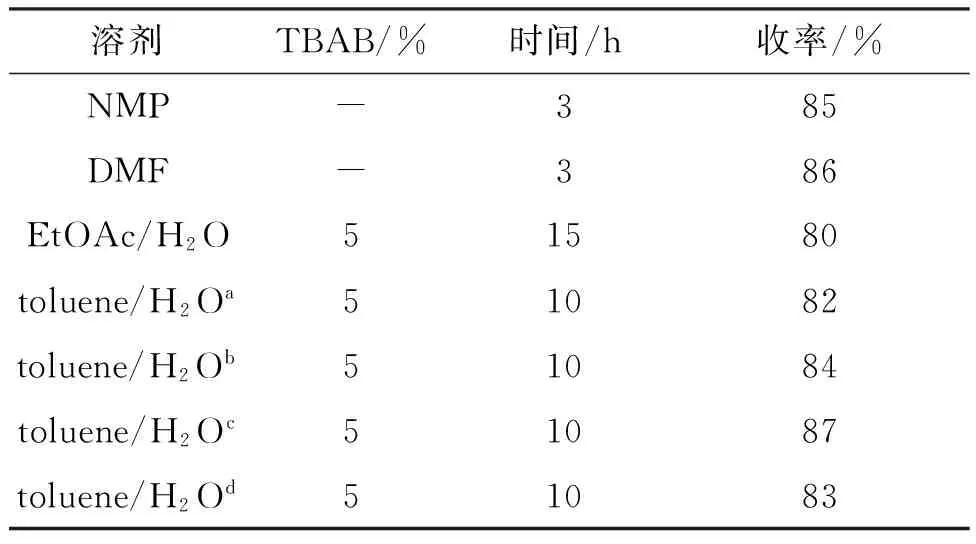
表1 溶剂对反应的影响
a~dV/V=1/1, 2/1, 3/1, 4/1。
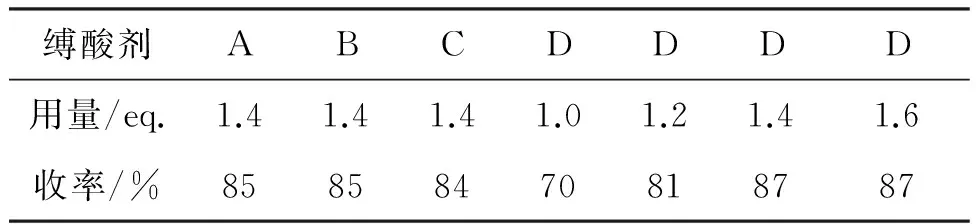
表2 缚酸剂对反应的影响*Table 2 Influence of the base on the reaction
*A~D依次为NaOH, KOH, Na2CO3, K2CO3。
表2为缚酸剂对反应的影响。由表2可知,在NaOH或KOH作用下,虽然收率较高,但杂质较多,产品质量较低。最终选择K2CO3作缚酸剂,用量为1.4 eq.,收率84%。
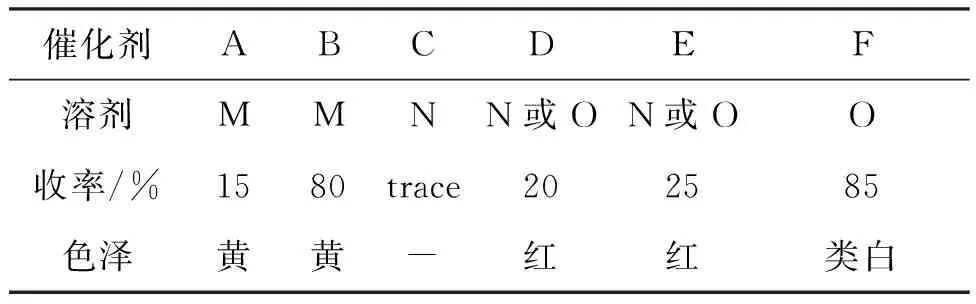
表3 催化剂对1收率的影响
aA~F依次为NH4Cl, Et3N·HCl, ZnO, ZnCl2, Zn(OTf)2, Zn(N3)2TMEDA;bM~O依次为甲苯,DMF,n-BuOH。
合成1时,有报道[13-14]采用ZnCl2或ZnBr2作催化剂进行催化。但实验中发现效果并不好。表3为催化剂对1收率的影响。由表3可知,用NH4Cl或Zn(OTf)2作催化剂,收率很低,副产物较多。以Et3N·HCl作催化剂,产品色泽较差。先将ZnCl2, TMEDA和NaN3预制为配合物,再与底物发生四唑化反应,效果较好。
由于反应所需温度较高,一般选用较高沸点的溶剂,结果见表4。由表4可知,正丁醇作溶剂时,产物的收率(85%)和色泽均较好。
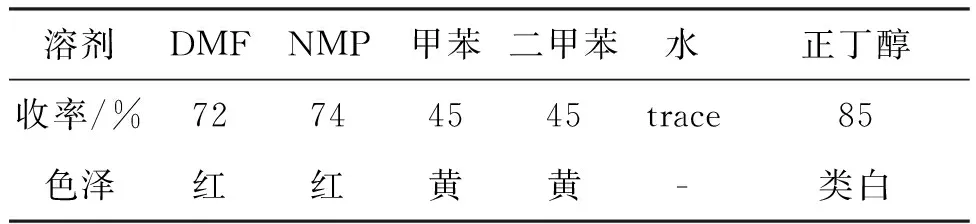
表4 溶剂对1收率的影响Table 4 Effect of solvents on the yield of 1
表5为TMEDA, ZnCl2和NaN3的用量对1收率的影响。由表5可知,当TMEDA, ZnCl2和NaN3的用量均为5的1.2 eq.时,收率最高(85%)。

表5 TMEDA, ZnCl2和NaN3用量对1收率的影响Table 5 Effect of the amount of TMEDA, ZnCl2 and NaN3 on the yield of 1
此外,我们还研究了反应时间和反应温度对1收率的影响。发现当反应时间超过40 h,反应温度超过110 ℃后,收率未显著提高,产品色泽变差。因此最佳反应温度为110 ℃,反应时间为40 h。
以2-氰基-4′-溴甲基联苯和2-丁基-4-氯-5-甲酰基-咪唑为原料,依次经N-烷基化、羟基化和四唑化反应合成了氯沙坦,总收率73.9%。该方法操作简单,避免使用危险性的试剂,产品色泽好,有一定的工业化前景。
[1] BRESCHI M C, CALDERONE V, DIGIACOMO M,etal. New no-releasingpharmacodynamic hybrids of losartan and its active metabolite:Design,synthesis,and biopharmacological properties[J].J Med Chem,2006,49(8):2628-2639.
[2] NIE Y Y, DA Y J, ZHENG H,etal. Synthesis and biological evaluation of novel potent angiotensin II receptor[J].Bioorg Med Chem,2012,20:2747-2761.
[3] JANG S Y, KIM S B, YUN S,etal. Method for Preparaing losartan:WO 2007133040[P].2007.
[4] YATENDRA K, VENKATA K R, MOHAN P,etal. Conversion of aromatic nitriles into tetrazoles:WO 2005051929[P].2005.
[5] MASAHIKO S, MASAKI N. Synthesis of angiotensin II receptor blockers by means of a catalytic system for C—H activation[J].J Org Chem,2011,76:10198-10206.
[6] 闫起强,郭拥政,谢苏豪,等. 氯沙坦的合成[J].合成化学,2010,18(1):83-85.
[7] 刘洋,朱小飞,俞建钧,等. 氯沙坦合成工艺改造[J].药学与临床研究,2011,19:89-90.
[8] 屠拥军,张毅,徐贤光. 一种合成氯沙坦及其中间体的方法:CN 102675294A[P].2012.
[9] VEERA R A, UDAYA B R, RAJENDIRAN C,etal. Proeess for the preparation of losartan:WO 2007026375[P].2007.
[10] MADASU S B, VEKARIYA N A, KOTESWARAMMA C,etal. An efficient,commercially viable,and safe process for preparation of losartan potassium,an angiotensin II receptor antagonist[J].Org Process Res Dev,2012,16:2025-2030.
[11] KIM, BYEONG C, AH N,etal. Novel zinc azide complex and a process for preparing tetrazole derivatives using the same:WO 2012148148[P].2012.
[12] SEDEKMEIER G. Process for the preparation of tetrazole derivatives from organo boron and organo aluminium azides :WO 2005014602[P].2005.
[13] 王亚平,郑国军,蔡刚华,等. 一种制备洛沙坦的方法:CN 1915990A[P].2007.
[14] DEMKO Z P, SHARPLESS K B. Preparation of 5-substituted 1H-tetrazoles from nitriles in water[J].J Org Chem,2001,66:7945-7950.
ProcessImprovementontheSynthesisofLosartan
ZANG Yong-jun1a, LIU Dong1a, HAN Bang-xing1b*
(a. School of Biology and Pharmaceutical Engineering; b.Center of Traditional Chinese Medicine Research ﹠ Development, West Anhui University, Luan 237012, China)
O623.42; O626.2
A
10.15952/j.cnki.cjsc.1005-1511.2017.10.17012
2017-01-16;
2017-08-07
安徽省教育厅一般项目(KJ103762015B16); 皖西学院自然科学基金青年基金资助项目(WXZR201613)
臧永军(1990-),男,汉族,安徽六安人,助教,主要从事药物合成的研究。 E-mail: zangyongjun@yeah.net
韩邦兴,教授, E-mail: hanbx1978@sina.com
猜你喜欢
健康体检与管理(2022年2期)2022-04-15 22:33:17
中国药学药品知识仓库(2022年5期)2022-04-11 21:25:52
石油石化绿色低碳(2018年5期)2018-03-20 04:41:21
石油炼制与化工(2016年6期)2016-04-06 22:54:21
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
合成化学(2015年2期)2016-01-17 09:04:08
合成化学(2015年2期)2016-01-17 09:03:13
中国卫生标准管理(2015年8期)2016-01-15 03:58:47
化工进展(2015年3期)2015-11-11 09:08:25
化学分析计量(2015年4期)2015-03-23 16:47:34
