
胎儿骨骼发育不良分子遗传学进展
2017-09-22 05:53:24综述吴青青审校
福建医科大学学报 2017年4期
刘 妍(综述), 吴青青, 于 松(审校)
胎儿骨骼发育不良分子遗传学进展
刘 妍(综述), 吴青青, 于 松(审校)
胎儿; 肌肉骨骼发育; 骨病医学; 遗传; 产前诊断
胎儿骨骼发育不良是包括骨骼生长发育异常的一系列疾病,由于骨骼分布广泛,由胎儿骨骼发育异常导致的骨骼畸形种类繁多,分类复杂,造成骨骼系统异常的原因多由遗传或环境因素(包括药物或机械作用)导致[1-3]。目前,几乎所有的胎儿骨骼异常都是靠产前超声诊断的[4-7]。然而,产前超声着重于区分致死性和非致死性骨发育不良,并不能对所有的骨骼系统畸形作出具体类型的判断,这也使得胎儿骨骼的分子遗传学诊断成为近年来胎儿骨骼异常产前诊断的重要手段[8-13]。笔者现就胎儿骨骼发育不良的产前诊断分类及分子遗传学研究进展做一综述,旨在为胎儿骨骼畸形的产前诊断提供帮助。
1 骨骼发育异常的分类及诊断
骨骼分布于全身各处,骨骼异常的表现形式多样、分类复杂。
1.1 根据骨骼发生的部位分类
1.1.1 全身性骨骼发育不良 原因多样,种类繁多,表现也多种多样,预后各不相同。对于全身性骨骼发育不良,超声主要将其诊断为致死性和非致死性骨骼发育不良。
李胜利等报道,区分致死性和非致死性骨发育不良的主要超声特征为[4]:(1)严重的四肢均匀短小畸形,四肢所有长骨长度均低于正常孕周平均值的4倍标准差。股骨长/腹围比值<0.16。(2)严重的胸部发育不良,常导致肺发育不良和胎儿死亡。主要诊断指标有:心胸比值>60%(除外心脏畸形),胸围/腹围比值<0.89,胸围低于正常平均孕周的第5百分位数。(3)某些特殊征象:如三角形头颅为致死性侏儒Ⅱ型特征表现,多发骨折为成骨不全Ⅰ型的特征。较常见的致死性骨发育不良包括:致死性侏儒、软骨发育不全、成骨不全Ⅱ型;少见的包括先天性低磷酸酶症、肢体屈曲症、骨骺点状发育不良、短肋多指综合征等。
非致死性骨发育不良较少见,发生率低于1/20 000,部分类型罕见。如:杂合子软骨发育不良,成骨不全Ⅰ、Ⅲ、Ⅳ型及窒息性胸廓发育不良等。
1.1.2 肢体局部畸形 肢体局部畸形包括横行肢体缺失、纵行肢体缺失、并腿畸形、裂手/足畸形、多指/趾、并指/趾等,肢体-体壁综合征(limb body wall complex,LBWC)和胎儿运动不能综合征(fetal akinesia deformationsequence,FADS)多伴典型的肢体局部畸形表现。
1.2 根据导致骨骼异常的原因分类
1.2.1 遗传性骨病 遗传性骨病是一大类具有广泛遗传异质性的骨软骨发育不良性疾病,大多是发病率很低的罕见病。按照2010年修订的骨发育异常国际分类标准,456种骨软骨发育不全按其分子遗传、影像及生化特点归纳为40组骨发育障碍,其中316种骨骼疾病与发现的226种骨骼致病基因的一种或多种相关(表1)[14]。
1.2.2 染色体异常所致的骨骼发育异常 不同种类的染色体异常,合并的骨骼畸形种类繁多,形式多样[15]。如,13-三体,18-三体,甚至在21-三体即唐氏综合症的患儿中也可以出现骨骼系统发育异常;其他一些常见的三倍体及常染色体的非整倍体也会合并不同类型的骨骼畸形。
1.2.3 其他因素导致 如羊膜带综合症及药物及原发性代谢疾病所致的骨骼发育异常。
1.3 根据肢体畸形的胚胎发生将骨骼系统畸形分为七大部分[16]。
1.3.1 部分形成失败 以肢芽形成失败为特征,又分横向与纵向两类。横向类涉及所有的先天性截肢,纵向类又分为挠侧尺侧,中央与中段形成失败,包括桡、尺骨部分或全部缺如。
1.3.2 部分分化失败 部分分化失败,有发育形成基本单位,但未分化形成正常组织结构,如骨性联合、并指、屈曲指等。
1.3.3 重复 部分肢芽或外胚层帽在早期受损,原始胚基形成裂隙,导致重复畸形,如多指、多趾畸形。
1.3.4 过度生长 所有肢体过度生长或局部肢体过度生长伴有神经脂肪血管浸润,如半侧肥大、巨指畸形。
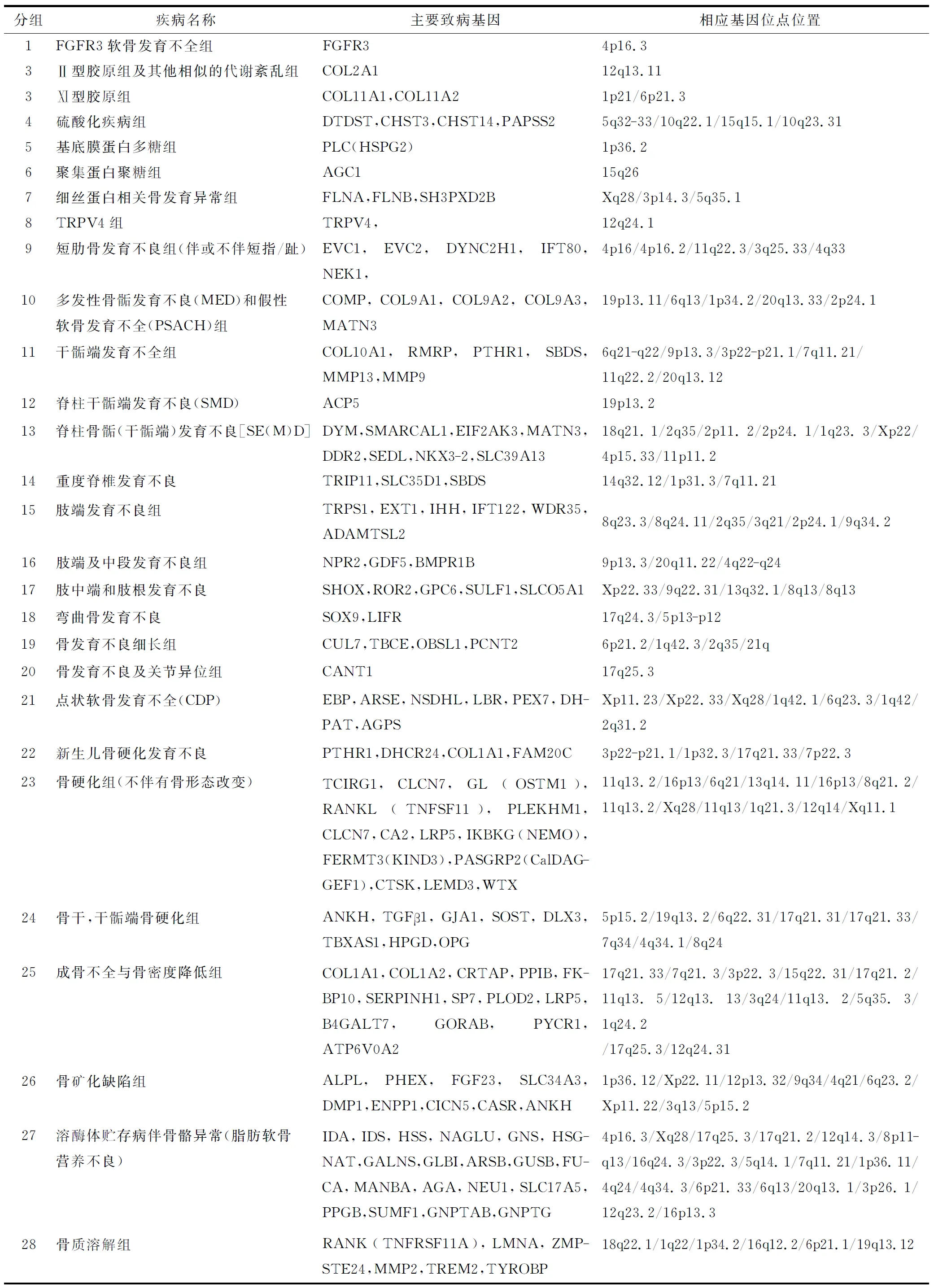
表1 《国际遗传性骨病分类标准(2010版)》疾病分组
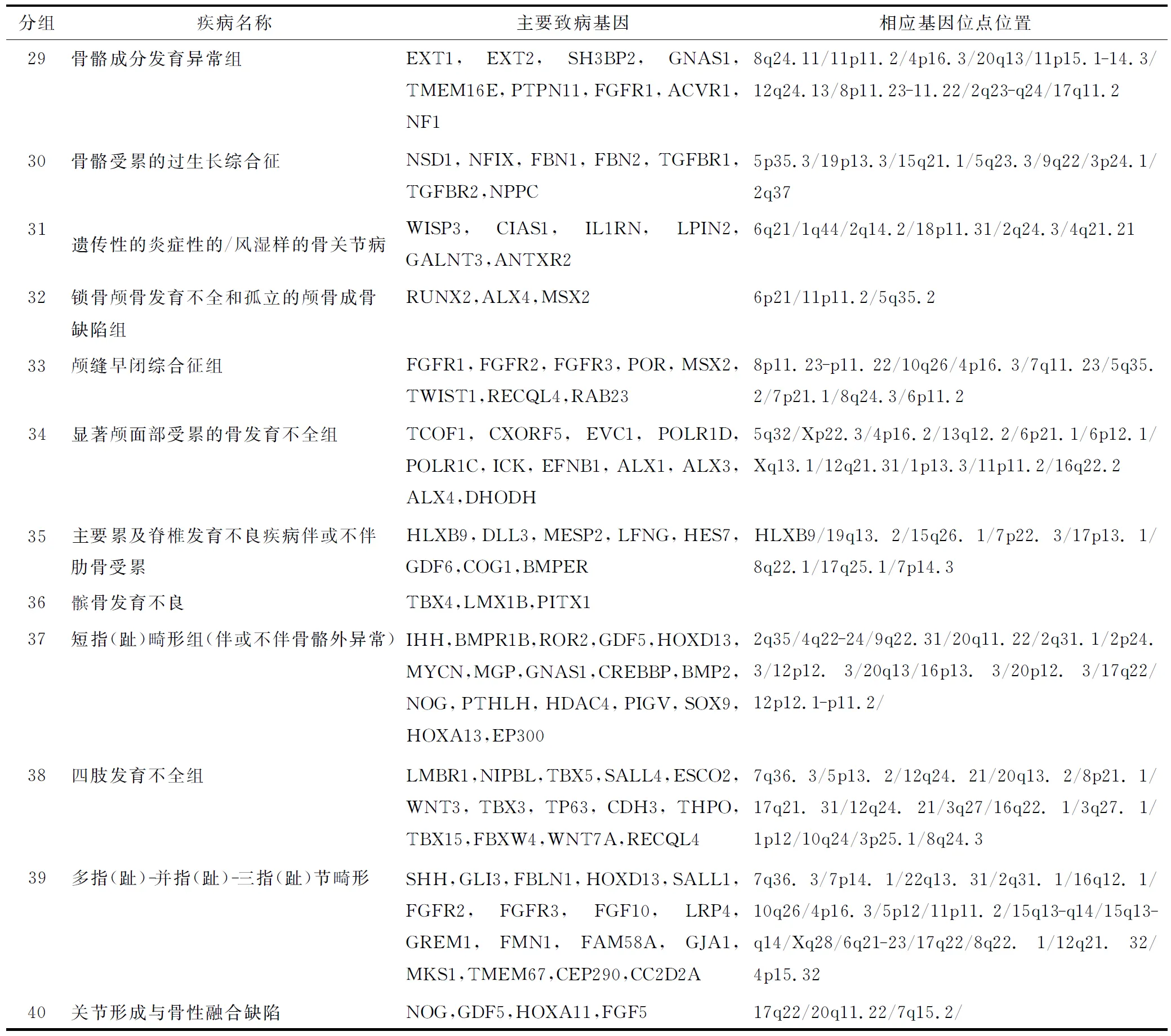
续表
注:主要参照 国际骨骼发育不良协会( International Skeletal Dysplasia Society)2010版国际遗传性骨病分类标准.
1.3.5 发育不全 指某一组织结构发育不完善,不完全,如短掌骨短、短指并指畸形。
1.3.6 先天性绞窄环综合征 与羊膜带综合征有关。羊膜带综合症可引起各种浅表软组织或深部骨骼发生坏死,严重者可表现为宫内截肢或截指,轻者只呈现软组织绞窄环挛缩带。
1.3.7 全身骨骼异常 指全身骨骼发育异常,包括染色体畸形、各类综合症、骨软骨发育异常等。
2 胎儿骨骼发育异常的病因研究
2.1 物理原因 如羊膜带综合症所致的物理方面的宫内截肢或截指。
2.2 染色体异常 可致不同种类的骨骼异常。如:13-三体的患儿会出现多指(趾)、马蹄内翻足、握拳手或手指交叠;18-三体会出现多指(趾)、肢体短小畸形、马蹄足内翻以及镰刀足;21-三体即唐氏综合症的患儿,可有短头、枕骨扁平、长骨短小、手指弯斜、并指畸形、手指粗短、草鞋足等表现。其他一些常见的三倍体也可以出现骨骼系统的异常,如手或脚三趾和四指的并指畸形、酒瓶样大腿。还有一些常染色体的非整倍体也会合并不同类型的骨骼畸形,如9-三体的婴儿有小头畸形、膝关节和肘关节错位,Turner综合症也会有股骨短小等表现。
2.3 分子遗传方面的基因诊断 不管是骨的发育、重塑、还是修复,都是在软骨细胞、成骨细胞、破骨细胞的协调作用下完成的。不同类型的细胞在时间及空间上形成一系列复杂的关系,如果突变造成基因表达的破坏则会引起相应的骨骼系统疾病,这包括了一些编码细胞、通信通路、重要的中介蛋白、基质蛋白及转录因子等。按照骨骼发育的遗传及分子生物学特点,骨病发育不良协会在2001年按致病机制将骨骼发育不良分为7组(表2)[17]。

表2 遗传性骨病的分子致病机制分类标准

续表
基于以上的致病基因对其进行的聚类分析如下:
2.3.1 细胞外基质结构蛋白缺陷 细胞外基质成分的基因包括胶原类基因COL1A1,COL1A2,COL2A1,COL11A1,COL11A2,COL9A1,COL9A2,COL9A3;肥大软骨细胞外基质的胶原类基因COL10A1;蛋白聚类基因MATN3,COMP,AGC1,HSPG2,GPC6;蛋白聚糖的糖链合成基因B3GAT3,EXT1/EXT2;蛋白聚糖硫酸化基因ATPSK2,CHST3,DTDST;及其他细胞外基质成分基因CMG2,FBN1,MGP。从骨骼异常表现分析,细胞外基质虽是骨骼的组成,但其相关基因突变却不仅造成骨基质或软骨基质的异常,而且能够影响细胞的增殖、分化,在整个成骨过程中扮演重要的角色。
2.3.2 胶原类基因突变疾病 Ⅰ型胶原突变常导致频发骨折、骨质疏松等骨骼异常表现;Ⅱ型胶原突变常导致身材矮小,骨骺骨化缺如等骨骼异常表现;Ⅸ型胶原突变常导致骨骺骨化延迟;Ⅺ型胶原突变可导致身材矮小,但受累较轻;Ⅹ型胶原突变常导致身材矮小。不管是软骨细胞分泌的Ⅱ型胶原还是肥大软骨细胞分泌的Ⅹ型胶原,在生长板的发育中均起到重要作用[18]。
2.3.3 蛋白聚糖及其修饰基因突变疾病 涉及蛋白聚糖及及糖基化、硫酸化修饰的基因突变,均导致编码蛋白的功能或者活性缺失,或滞留于内质网,导致身材矮小、关节异常。累及部位多数为四肢和躯干骨,较少累及颅面部;糖基化或硫酸化修饰的基因突变还较常累及手部、足部、指(趾)骨。
殖民时代的国际经贸交往与其说是合作,不如说是资源掠夺。然而,即便宗主国以剥削殖民地资源和输出资本为目的进行经贸往来或者说殖民扩张,他们也注意到了消除混乱、确保资源的获取需要法律规则,只不过此时的殖民地附属于宗主国,因此无所谓通过国际法层面消除混乱的问题,宗主国直接采取法律强制输出的方式使殖民地的法律与宗主国趋同,就可以实现利用法律规则控制地区冲突,消除社会混乱的目的。
在蛋白聚糖的基因中,HSPG2突变的DDSH疾病的症状最严重,表现为新生儿致死性侏儒病,导致较罕见的椎体分节不均的表型[19]。MATN3,COMP,AGC1三者滞留内质网时均会造成骨关节炎,进而导致关节变形;ATPSK2,COMP突变导致的疾病还会表现为骨骺骨化延迟;B3GAT3与EXT1/EXT2都是蛋白聚糖的糖基化蛋白,两者的功能缺失突变均会造成骨化中心增多的现象。但EXT1/EXT2突变导致较罕见的多发性良性骨肿瘤,而B3GAT3突变却会导致身材矮小、关节脱位、骨质疏松等。
2.3.4 信号通路 骨相关细胞信号传导是指除遗传信息传递的中心法则外,细胞环境信息传递的分子途径。(1)成纤维细胞因子(fibroblast growth factor, FGF)通路:FGF通路共包括18种以上的配体和4种以上的受体,多数为FGFR突变。FGFR的突变多数为功能激活突变,可导致骨骼系统受累较严重的软骨发育不全、致死性发育不全、季肋发育不全等疾病,而功能缺失突变导致骨骼受累较轻的泪管-耳-齿-指综合征等疾病。突变区域分布于各自的胞外区或酪氨酸激酶区,不同位点的突变可导致不同的疾病,如FGFR1,FGFR3突变会影响肢体的发育导致侏儒、长骨弯曲的表型,而FGFR2的作用不明显。FGF9,FGF10,FGF23三种配体的突变表型各异,不同的位点突变也会导致同样表型。如FGF10,FGFR2,FGFR3的功能缺失突变均可造成拇指的三指节畸形,说明三者在拇指发育中缺一不可,FGF10有可能同时通过两种受体发挥作用。而FGF23突变导致低磷酸血症的佝偻病表型,说明FGF23主要通过调节磷酸水平而调节骨代谢。(2)甲状旁腺素 (parathyroid hormone, PTH)通路。PTH通路紊乱导致的骨骼异常多累及长骨,突变类型各异,导致的表型各异。PTH信号通路在调节生长板的软骨细胞增殖、生长、分化过程中具有明显的剂量效应,只有适当的PTH信号通路,才能够保证正常的成长过程。甲状旁腺相关蛋白PTHR(PTH-related protein)的突变还会导致骨骺骨化延迟、骨骼过早成熟等异常现象,说明PTH通路也参与软骨内成骨的固化环节的调节。(3)转化生长因子β(transformig growth factor beta, TGFβ)的信号通路。TGFβ包括3种亚型:TGFβ1、TGFβ2和TGFβ3。TGFβ通路增强导致四肢短的表型、TGFβ1信号增强可导致骨皮质增厚的表型,说明TGFβ在骨发育中的作用多样,不同亚型之间可能存在差异。(4)骨形态发生蛋白(bone morphogenetic protein, BMP)信号通路。涉及BMP信号通路的突变基因包括配体GDF-5,GDF-6,受体BMPR1B,ACVR-1和通路抑制分子NOG,LEMD3。骨骼异常表现主要为椎体、远端肢体关节异常融合、某些骨骼部件缺失、软组织异位骨化等。GDF5的功能增强突变或缺失突变都会造成肢体关节的异常融合、某些骨骼部件的缺失(例如股骨、腓骨等)、短指等症状。同样是BMP受体,与BMPR1B突变不同的是,ACVR-1的突变却导致软组织的异位骨化,并为软骨内成骨形式,说明ACVR-1介导的BMP通路可主导成纤维细胞向软骨细胞分化继而骨化的过程。NOG与LEMD3同为BMP通路的抑制因子,但两者突变表型差距较大,说明BMP介导的通路多样性,且BMP通路可能受到各方面复杂的调节作用。(5)WNT信号通路。主要涉及通路中的配体分子WNT3,WNT7A,WNT5A,抑制分子WTX,SOST,受体分子LRP5,ROR2。与其他信号通路不同的是,WNT信号通路分子的突变在骨骼异常表现方面不仅累及四肢骨、中轴骨,颅盖骨也表现出明显的发育异常现象,主要表现为四肢的发育缺陷或缺如及长骨、颅盖骨的骨量异常变化[20]。LRP5和ROR2的功能缺失突变会导致身材矮小的症状;WTX和SOST的功能缺失突变会表现为长骨、颅盖骨的骨量增加;而SOST的功能缺失突变还会导致巨人症的表型;WNT3,WNT7A,WNT5A三种配体分子中,WNT3/WNT7A突变后会导致严重的四肢发育缺陷,而WNT5A突变的症状则相对较轻,主要表现为身材矮小,牙齿异常。(6)NOTCH信号通路。NOTCH信号在骨骼系统中,对体节生成、成骨细胞和破骨细胞的分化、成熟具有复杂的调控作用。NOTCH信号突变可引起脊柱肋骨发育不全、脊柱胸廓发育不全、Alagille综合征和短指/趾等骨骼疾病[21-22]。(7)Hedgehog信号通路。涉及Hedgehog信号通路的疾病主要为配体分子SHH,IHH,转录因子Gli3以及调控因子Lmbr1,KIF7突变引起,这些疾病的骨骼异常主要表现为并指(趾)、短指(趾)、多指(趾)。IHH突变的疾病主要表现为短指(趾)、骨骺生长板融合过早造成的短肢等;而SHH,Gli3以及调控因子Lmbr1,KIF7突变的疾病主要表现为多指(趾)。IHH突变后既可以导致表型轻微的短指(趾)症状,又可以导致表型更为严重的伴短指(趾)的股骨头发育不良疾病,说明IHH的不同结构域或剂量具有一定的组织部位特异性[23]。
2.3.5 细胞骨架 细胞骨架相关基因的突变,可累及骨骼、皮肤、血管、心脏等多个器官,患病严重程度各异。FLNA,FLNB突变可致骨骼矿化缺陷;纤毛、鞭毛形成的相关基因突变主要表现为短肋、短肢、颅面异常、多指(趾)等;微管、纺锤体形成基因突变与生长迟缓、皮质骨增厚、颅面异常相关;细胞核骨架基因突变的疾病,主要表现为生长发育迟缓、下颌骨发育不良,远端指、趾骨、锁骨进行性骨溶解。
2.3.6 钙-磷代谢基因 钙-磷代谢相关基因突变疾病主要累及骨骼、肾、甲状腺等,以骨骼异常表现最明显。
2.3.7 转录因子 目前已知有很多转录因子在软骨生成和软骨内骨化阶段对骨骼的发生进行了调控,如SHOXY,SOX9,TBX4,HOXD13,RUNX2,MSX2等。作为信号通路的终端作用分子,转录因子突变后的骨骼异常主要表现为某些骨骼元件的发育不良和缺如等,多数以颅面骨畸形、指(趾)骨缺如、并指(趾)及多指(趾)为主,个别疾病以长骨发育缺陷为主[24]。如BX4,HOXA13,SALL4,TBX3,TP63,HOXD13突变主要导致手足异常;TBX4,LMX1B可导致特有的髌骨发育不全;SOX9,DLX3,SHOX/SHOXY突变主要累及长骨,以挠骨、尺骨受累为主;RUNX2,MSX2,TWIST1ALX1-4基因突变所致疾病以颅面骨受累为主[25]。
2.3.8 脂类代谢 脂代谢相关基因主要为胆固醇、磷脂合成相关基因,这些基因的突变主要导致皮肤、骨骼、毛发、神经系统异常,导致的骨骼异常表现除了明显的某些骨骼部件发育不全外,主要为骨骼矿化异常。
2.3.9 破骨细胞相关基因 与破骨细胞分化、功能相关的基因主要包括重要信号通路RANKL/RANK;与吸收陷窝的酸化有关的TCIRG1,OSTM1,CLCN7,PLEKHM1,CAII,以及吸收骨质主要酶类TRAP。
总之,遗传性骨病的分子遗传学研究进展迅速,研究应该结合骨骼生物学和骨软骨发育不良的分子遗传学研究建立更加高效的检测方法,为遗传性骨病的诊断、产前诊断及基因治疗奠定基础。骨骼系统病种庞大,表现形式复杂多变,如何能够提高胎儿骨骼发育异常的产前诊断正确率,寻找致病原因,并对其进行相应的遗传咨询及再发风险评估,指导再次妊娠,是产科及超声科医生共同面临的一个巨大的挑战。
[1] Cheon J E.Genetic Skeletal Disorders[M].RadiologyIllustrated:PediatricRadiology.Springer Berlin Heidelberg,2014:887-911.
[2] Shah I P,Varghese B,Fernandes J A. Skeletal dysplasia[J].Surgery, 2017,35(1):52-61.
[3] Sewell M D,Chahal A,Al-Hadithy N,etal. Genetic skeletal dysplasias:A guide to diagnosis and management[J].JBack&MusculoskeletalRehabilitation,2015, 28(3):575-590.
[4] 李胜利.胎儿肢体畸形产前超声诊断及预后[J].中国实用妇科与产科杂志, 2007,23(5):399-400.
[5] Barkova E,Mohan U,Chitayat D,etal.Fetal skeletal dysplasias in a tertiary care center: radiology, pathology,and molecular analysis of 112 cases[J].ClinicalGenetics,2015,87(4):330-337.
[6] Cho I,Shim J Y,Kim G H,etal.Thanatophoric dysplasia in a dichorionic twin confirmed by genetic analysis at the early second trimester: A case report and literature review[J].Obstetrics&GynecologyScience,2014,57(2):151-154.
[7] Berceanu C,Gheonea I A,Vlǎdǎreanu S,etal.Ultrasound and MRI comprehensive approach in prenatal diagnosis of fetal osteochondrodysplasias[J].MedicalUltrasonography,2017,19(1):66.
[8] Nelson D B, Dashe J S, Mcintire D D,etal.Fetal skeletal dysplasias: sonographic indices associated with adverse outcomes[J].JournalofUltrasoundinMedicine, 2014,33(6):1085-1090.
[9] Yeh P,Saeed F,Paramasivam G,etal.Accuracy of prenatal diagnosis and prediction of lethality for fetal skeletal dysplasias[J].PrenatDiagn, 2011,31(5):515-518.
[10] Toru H S,Nur B G,Sanhal C Y,etal.Perinatal diagnostic approach to fetal skeletal dysplasias:six years experience of a tertiary center[J].FetalandPediatricPathology,2015,34(5):287.
[11] Shaffer L G, Rosenfeld1 J A,Dabell M P,etal.Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound[J].PrenatalDiagnosis, 2012,32,986-995.
[12] Polla D L,Cardoso M T,Silva M C,etal.Use of targeted exome sequencing for molecular diagnosis of skeletal disorders[J].PlosOne, 2015,10(9):e0138314.
[13] Sperber S,Spector E.FibroblastGrowthFactorReceptorandRelatedSkeletalDisorders[M].Molecular Pathology in Clinical Practice. Springer International Publishing, 2016.
[14] Bonafe L,Cormier Daire V,Hall C,etal.Nosology and classification of genetic skeletal disorders: 2015 revision[J].AmericanJMedicalGenetics(Part A),2015,167A(12):2869.
[15] 熊 雯,罗 红,安绍宇,等.胎儿骨骼系统异常与染色体异常的相关性分析[J].中华医学超声杂志(电子版),2015(2):148-151.
[16] 李胜利.胎儿畸形产前超声诊断学[M].北京:人民军医出版社,2004.
[17] Supertifurga A,Bonafé L,Rimoin D L,etal. Molecular-pathogenetic classification of genetic disorders of the skeleton[J].AmericanJMedicalGenetics, 2001, 106(4):282-293.
[18] 郑 谦,张筱薇. X型胶原蛋白与骨骼生长发育关系的研究进展[J].国外医学(口腔医学分册), 2002(2):73-75.
[19] Lowe D A,Leporibui N,Fomin P V,etal. Deficiency in Perlecan/HSPG2 during bone development enhances osteogenesis and decreases quality of adult bone in mice[J].CalcifiedTissueInternational,2014,95(1):29.
[20] 陆 诚. Wnt信号通路在软骨发育中的功能研究[D].上海:上海交通大学,2013.
[21] Yorgan T,Vollersen N,Riedel C,etal. Osteoblast-specific Notch2 inactivation causes increased trabecular bone mass at specific sites of the appendicular skeleton[J].Bone, 2016,87:136-146.
[22] 左宇志,刘振磊,刘 森,等. Notch信号通路与骨骼系统的研究进展[J]. 中华实验外科杂志, 2015,32(12):3238-3240.
[23] 陈祥和,李世昌,严伟良,等. Hedgehog信号通路对成骨细胞分化和骨形成的影响及不同方式运动的调控[J]. 北京体育大学学报, 2015(11):59-64.
[24] 孙冬梅. 决定骨骼发育的转录因子的转录调控机制的研究[D].长春:东北师范大学,2009.
[25] 吕学敏,杨庆铭,邓廉夫. 调控软骨生成和软骨内骨化的转录因子及其相关机制研究进展[J].中国矫形外科杂志,2004,12(z2):268-270.
(编辑:常志卫)
2017-03-20
首都医科大学 附属北京妇产医院产科,北京 100026
刘 妍,女,主治医师,医学硕士
R681; R714.53; R719.8
: A
: 1672-4194(2017)04-0267-08
猜你喜欢
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年5期)2021-08-24 07:06:28
中国临床医学影像杂志(2019年5期)2019-08-27 02:48:00
现代园艺(2017年21期)2018-01-03 06:41:32
小布老虎(2017年1期)2017-07-18 10:57:27
罕少疾病杂志(2016年4期)2016-03-11 16:34:41
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
少年科学(2009年12期)2009-07-07 07:05:10
