
基于微小RNA架构的新型随机短发夹RNA文库构建
2017-09-08 06:33卜萌萌郭思超闫雪静陈梅红
中国医学科学院学报 2017年4期
郭 倩,朱 宁,卜萌萌,郭思超,闫雪静,陈 倩,陈梅红
中国医学科学院基础医学研究所 北京协和医学院基础医学院生物化学与分子生物学系 医学分子生物学国家重点实验室,北京 100005
基于微小RNA架构的新型随机短发夹RNA文库构建
郭 倩,朱 宁,卜萌萌,郭思超,闫雪静,陈 倩,陈梅红
中国医学科学院基础医学研究所 北京协和医学院基础医学院生物化学与分子生物学系 医学分子生物学国家重点实验室,北京 100005
目的 构建高效发夹状随机短发夹RNA(shRNA)文库。方法 构建由pol Ⅱ启动子(CMV)驱动的增强绿色荧光蛋白(EGFP)下游连接shRNA的表达载体,转染细胞后收取蛋白,用Western blot和荧光素酶报告基因系统检测该载体表达的shRNA对靶基因表达的抑制效率。在由CMV启动的EGFP下游连接shRNA的表达载体基础上,构建EGFP下游连接基于微小RNA(miRNA)架构的shRNA表达载体,转染细胞后收取RNA,用实时定量荧光PCR 法检测该载体表达的shRNA对靶基因表达的抑制效果。基于EGFP下游连接基于miRNA架构的shRNA表达载体实时定量荧光PCR的检测结果,构建基于miRNA架构的大容量随机shRNA文库。结果 由pol Ⅱ启动子(CMV)驱动发夹状shRNA转录时,shRNA要位于一个大的转录本之后才能被有效转录。基于miRNA架构环境可提高shRNA的干扰效率。构建了一个容量为1.8×107的基于miRNA架构的大容量随机shRNA文库。结论 构建了一个大容量的新型随机shRNA文库。
微小RNA;短发夹RNA;随机文库
RNA干扰(RNA interference,RNAi)已经成为抑制基因表达的常用方法,对于载体表达的小干扰RNA(small interfering RNA,siRNA),siRNA表达的高效性和siRNA的特异性是决定siRNA干扰效率的主要因素[1- 6]。微小RNA(microRNA,miRNA)是内源性非编码小RNA,miRNA基因在核内被RNA多聚酶pol Ⅱ转录成原miRNA,由核酸内切酶Drosha、DGCR8复合物加工形成前体miRNA(miRNA precursor,pre-miRNA),pre-miRNA进一步在细胞质中被Dicer、TRBP复合物剪切形成成熟的miRNA,和蛋白因子一起形成RNA诱导的沉默复合物,介导与miRNA有同源序列的靶mRNA的表达抑制。对miRNA成熟过程的研究使人们对于小RNA的表达及特异性有了更深的认识[1,3]。在RNAi技术中,除了siRNA外,载体表达的短发夹RNA(short hairpin RNA,shRNA)常用于稳定敲低靶基因。随着技术的不断发展,针对全基因组的大规模shRNA干扰文库得以实现,广泛用于筛选生物过程中发挥关键作用的基因。与针对已知基因的文库相比,随机shRNA具有基因覆盖面更广的优点,所以构建一个高效的大容量shRNA随机文库具有重要的实用价值[3]。现有的几个随机siRNA/shRNA文库在文库大小及有效shRNA含量方面都还不尽人意[7- 8]。本研究旨在构建一种高效shRNA随机文库的新策略。
材料和方法
材料 H1和U6启动子驱动的siRNA表达载体pCM-H1U6由本实验室构建[7]。pcDNA3.1、pLenti6.3-V5-TOPO载体购自Invitrogen公司。文库构建引物:A 引物:GTCCGAGACAGTAGGCACCGCC;B 引物:CTCGAGAAGGTATATTGCTGTTGACAGTGAGCGACGCC;C 引物:CAGTAGGCACCGAATTCTAGCCCCTTGAAGTCCGAGG 均由Invitrogen公司合成。shNC-QT1是阴性对照shRNA,序列由Dharmacon公司设计。shhhnRNPA0是针对人hnRNPA0基因设计的shRNA,shhhnRNPA2B1是针对人hnRNPA2B1基因设计的shRNA,将它们置于miRNA- 30架构中并位于增强绿色荧光蛋白(enhanced green fluorescent protein,EGFP)下游,克隆入pc-DNA3.1载体中,构建成pcDNA3.1-EGFP-mir-shNC-QT1、pcDNA3.1-EGFP-mir-shhnRNPA0、pcDNA3.1-EGFP-mir-shhnRNPA2B1质粒;shhnRNPA0正义链序列:GGCGGTCGCAGTAATAGTGGA;shhnRNPA2B1正义链序列:GGAACAGTTCCGTAAGCTCTT;将针对人NOV基因和萤火虫荧光素酶报告基因的shRNA克隆入pLenti6.3-V5-TOPO载体构建成pLenti6.3-shNOV、plenti6.3-EGFP-shNOV、pLenti6.3-shFL867、plenti6.3-EGFP-sh FL867质粒。shNOV 正义链序列:GCACCAAGAAGTCACTCAA;shFL867 正义链序列:CCCTATTCTCCTTCTTCGC。限制性内切酶、T4 DNA连接酶、DNA 聚合酶Ⅰ(Klenow酶)购自NEB公司,PrimeSTAR®HS DNA Polymerase、电转化感受态细胞(E.coliDH5α Electro-Cells)、RNAiso plus、DL 2000 DNA相对分子质量标准购自Takara公司,QIAquick®Gel Extraction kit、QIAquick®Nucleotide Removal kit购自QIAGEN公司,EndoFree Plasmid Midi Kit、UltraSYBR Mixture购自北京康为世纪生物科技有限公司。DMEM培养基购自Thermo scientific公司,胎牛血清购自北京四季青公司,Lipofectamine 2000购自Invitrogen公司,GoScriptTMReverse Transcriptase、Dual-Luciferase Reporter Assay System购自Promega公司。HEK293T、MG63细胞购自中国医学科学院基础医学研究所细胞中心。
细胞转染 HEK293T细胞、MG63细胞在转染前一天按细胞数1×105个/孔铺24孔板中,次日转染时,待细胞混合度达到80%~90%时,按Lipofectamine 2000转染试剂说明书操作,将质粒转染入细胞。转染后48 h后收取细胞。
RNA提取和实时定量PCR 收集细胞后,用1×PBS重悬细胞清洗3次,弃上清后加入1 ml RNAiso plus,按照说明书提取RNA,采用GoScriptTMReverse-Transcriptase将提取的RNA反转录成cDNA。用UltraSYBR Mixture进行荧光定量实时定量PCR检测,使用Bio-Rad CFX96荧光定量PCR仪,以beta-actin为内参,采用相对定量方法分析实验数据。
Western blot和荧光素酶检测 收集细胞,加入细胞裂解液、蛋白酶抑制剂、磷酸酶抑制剂,重悬混匀后于冰上震荡15 min,4℃ 12 000×g 离心15 min,吸取上清于另一干净的离心管中。Nanodrop测定蛋白浓度,30 μg蛋白/孔,10%聚丙烯酰胺凝胶电泳,半干转膜法将蛋白转移到聚偏氟乙烯膜上,5%牛血清蛋白室温封闭2 h,按照合适的比例用5%牛血清蛋白稀释一抗,4℃孵育过夜,TBST漂洗液漂洗3次,每次10 min,再用辣根过氧化物酶标记二抗孵育2 h,TBST漂洗液漂洗3次,每次10 min,于暗室用ECL发光试剂盒曝光X片。用Dual-Luciferase Reporter Assay System试剂盒检测共转染的萤火虫和海肾荧光素酶活性。
表达质粒构建 采用miRNA miR- 30的原miRNA序列架构[1],用目的shRNA序列替换miR- 30成熟序列,保留miR- 30成熟序列的5’和3’外侧序列和环序列,形成基于miRNA架构的shRNA表达框,将其克隆入pcDNA3.1载体中EGFP的下游,构建成与大转录本EGFP共表达的基于miRNA架构的shRNA表达质粒,其中miR- 30成熟序列的5’外侧序列为:5’ GACTTCTTAACCCAACAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG 3’,3’外侧序列为:5’ TGCCTACTGCCTCGGACTTCAAGGGGCTA 3’,环序列为:5’ TAGTGAAGCCACAGATGTA 3’。
统计学处理 采用SPSS 13.0统计软件,数据以均数±标准差表示,组间比较采用方差分析LSD检验,P<0.05为差异有统计学意义。
结 果
pol Ⅱ 启动子驱动大转录本之后的shRNA转录 构建了shRNA直接位于pol Ⅱ启动子(CMV)下游的pLenti6.3-shNOV、pLenti6.3-shFL867质粒和shRNA位于EGFP之后的plenti6.3-EGFP-shNOV、pLenti6.3-EGFP-shFL867质粒。MG63细胞瞬时转染质粒,其中shFL867表达质粒与萤火虫和海肾荧光素酶报告基因表达载体共转染细胞,转染48 h后收取细胞,Western blot结果显示,转染plenti6.3-EGFP-shNOV后,靶NOV蛋白水平明显降低,而转染pLenti6.3-shNOV组的NOV蛋白水平无变化(图1A);转染plenti6.3-EGFP-shFL867后,靶萤火虫荧光素酶蛋白活性明显降低,而转染pLenti6.3-shFL867组的萤火虫荧光素酶水平无变化(图1B)。
基于miRNA架构的shRNA干扰效率 将EGFP共表达的基于miRNA架构的shRNA表达质粒瞬时转染HEK293T细胞,48 h后提取细胞RNA,反转录成cDNA进行实时荧光定量PCR检测,结果显示:相对于H1和U6启动子驱动的siRNA表达载体pCM-H1U6表达的siRNA,位于一个大的转录本之后的shRNA对靶基因的抑制效率明显增加,由位于大的转录本之后的基于miRNA架构的shRNA表达框表达的shRNA对靶基因表达的抑制程度进一步提高(图2)。

图 1 与大转录本共表达的shRNA对靶基因的抑制效率
Fig 1 Target gene inhibition efficiency of shRNA co-expressed with large transcript

miRNA:微小RNA;与shRNA阴性对照shNC-OT1比较,aP<0.05,bP<0.01
miRNA:microRNA;aP<0.05,bP<0.01 compared with shRNA negative control shNC-OT1
A.HEK293T细胞转染shRNA表达质粒后靶基因hnRNPA0的mRNA水平;B.HEK293T细胞转染shRNA表达质粒后靶基因hnRNPA2B1的mRNA水平
A.mRNA level of hnRNPA0 in HEK293T cells transfected with shRNA expression plasmids;B.mRNA level of hnRNPA2B1 in HEK293T cells transfected with shRNA expression plasmids
图 2 基于miRNA架构的载体表达的shRNA对靶基因的抑制效率
Fig 2 Target gene inhibition efficiency of shRNA expressed by miRNA context-based expression vector
基于miRNA架构的大容量随机shRNA文库的构建 基于上述实时定量荧光PCR结果,构建基于miRNA架构的大容量随机shRNA文库(图3)。siRNA的中间部分序列在其干扰作用的特异性上发挥主要作用,因此在随机shRNA文库中,shRNA中间的16个碱基被设计为随机序列,两端为固定序列。首先化学合成含随机shRNA序列的寡聚核苷酸,将其变性退火形成一个半发夹结构,用Klenow酶将其补平,形成一个完整的发夹结构(图4A)。以A为引物,用PrimeSTAR®HS DNA聚合酶将发夹结构打开成为双链结构(图4B)。以双链结构为模板,以B和C为引物,通过PCR合成两端带有miRNA架构序列、拼接序列以及EcoR I酶切位点的双链片段(图4C)。以带有miRNA架构序列、拼接序列以及EcoR I酶切位点的双链片段为模板,PCR合成两端带有拼接序列以及Apa1酶切位点的EGFP片段。用PCR将上述两片段拼接成EGFP下游带有基于miRNA架构的shRNA表达框的片段EGFP-mir-sh16N(图4D)。将EGFP-mir-sh16N片段克隆在pcDNA3.1的Apa1和EcoR1酶切位点之间。将酶切后的7μg Apa1和EcoR1 pcDNA3.1载体和1∶3摩尔比的EGFP-mir-sh16N片段连接,连接产物电转化E.coliDH5α Electro-Cells,SOC培养基复苏后全部加入到500 ml的LB液体培养基中,从中取出10 μl涂布具有Amp+LB的固体培养基,长出约900个单克隆,从中挑取10个单克隆质粒酶切鉴定并测序,计算文库的大小。酶切鉴定显示:挑取的10个单克隆中,有9个酶切鉴定有插段(图5),测序结果显示其中1、3、4、5、7号克隆只有部分shRNA 正义链序列,缺失环和反义链序列,2号克隆未插入片段,6、8、9、10号克隆序列结构正确,且其shRNA序列各不相同,符合随机shRNA序列特征(表1),根据转化所得的总菌落数、酶切鉴定和测序结果计算文库的大小,计算方法:实际文库大小=转化所得的总菌落数×酶切鉴定含插段质粒的比例×含插段质粒的测序正确比例,本研究构建的实际文库大小=900÷10×500000×90%×4÷9=1.8×107。

图 3 基于miRNA架构的随机shRNA文库构建策略图
Fig 3 The strategy of constructing miRNA context-based random shRNA library

M:DL2000 DNA相对分子质量标准;1:半发夹结构寡聚核苷酸;2:补平后的发夹结构;3:发夹结构打开后的双链结构;4:带有miRNA骨架序列、拼接序列以及酶切位点的双链片段;5:带有拼接序列以及酶切位点的EGFP片段;6:拼接后的EGFP下游带有含miRNA架构的shRNA编码序列的片段EGFP-mir-sh16N
M:DL2000 DNA ladder;1:semi-hairpin oligo;2:hairpin oligo blunted;3:double-strand fragment opened from hairpin oligo;4:double-strand fragment containing miRNA context,splicing sequence and restriction enzyme sites;5:EGFP fragment followed by splicing sequence and restriction enzyme sites;6:fragment EGFP-mir-sh16N with EGFP followed by shRNA coding sequence embedded in miRNA context
A.发夹结构的寡聚核苷酸形成;B.发夹结构打开成为双链结构;C.扩增带有miRNA 骨架序列、拼接序列以及酶切位点的双链片段;D.拼接成EGFP下游带有含miRNA架构的shRNA编码序列的片段
A.generation of hairpin oligo;B.opening of hairpin oligo to generate double-strand fragment;C.amplifying the double-strand fragment containing miRNA context,splicing sequence and restriction enzyme sites;D.splicing the fragment with EGFP followed by shRNA coding sequence embedded in miRNA context
图 4 构建文库克隆过程中的电泳鉴定
Fig 4 Electrophoresis identification of the products in cloning
讨 论
近年来,RNAi技术在功能基因组学中显示出广阔的应用前景,可以利用RNA干扰进行大规模、高通量的功能基因筛查,由此出现了针对基因组范围内大量基因的 siRNA 汇总的RNA干扰文库,为大规模的基因功能分析和研究提供了有力的工具。

M:DL2000 DNA相对分子质量标准;1~10:文库质粒用EcoR1、Apa1双酶切鉴定
M:DL2000 DNA ladder;1- 10:plasmids from the library,digested with EcoR1 and Apa1
图 5 基于miRNA架构的shRNA随机文库质粒的酶切鉴定
Fig 5 Identification of the inserts in miRNA context-based random shRNA library plasmids
现有的几个随机siRNA/shRNA文库在文库大小及有效shRNA含量方面都还不尽人意[7- 8]。与pol Ⅲ启动子(H1、U6)驱动的siRNA表达相比,pol Ⅱ启动子驱动的shRNA表达具有能实现可诱导性表达及组织特异性表达的优点[2]。近年对于miRNA的剪切成熟过程有了较深入的认识,有研究表明,将合成的shRNA嵌入到内源的miRNA架构中可以高效诱导RNAi效应,由于嵌入到内源miRNA架构中的shRNA可以被精确剪切处理,从而提高shRNA的特异性,减少脱靶效应[1- 2],由此发展出将人工设计的shRNA置于miRNA架构中以提高shRNA的表达水平和干扰效率的shRNA设计新思路,本研究在这些工作的基础上,建立了一种构建高效shRNA随机文库的新策略。
本研究显示pol Ⅱ启动子转录shRNA时,shRNA必须置于一个大的转录本之后才可以被有效地转录。据此构建了pol Ⅱ启动子驱动的、shRNA位于大转录本EGFP下游并置于内源性miR- 30的miRNA架构中的新型大容量随机shRNA文库。测序鉴定结果显示,该文库具有1.8×107的实际容量。在测序的9个文库质粒中,4个具有正确的miRNA架构和随机shRNA序列特征,5个缺失了环和反义链序列,只有部分正义链序列,可能是化学合成的含随机shRNA序列的寡聚核苷酸序列较长,合成过程中出错造成的,如果提高化学合成寡聚核苷酸的正确性,文库的容量还可以进一步提高。
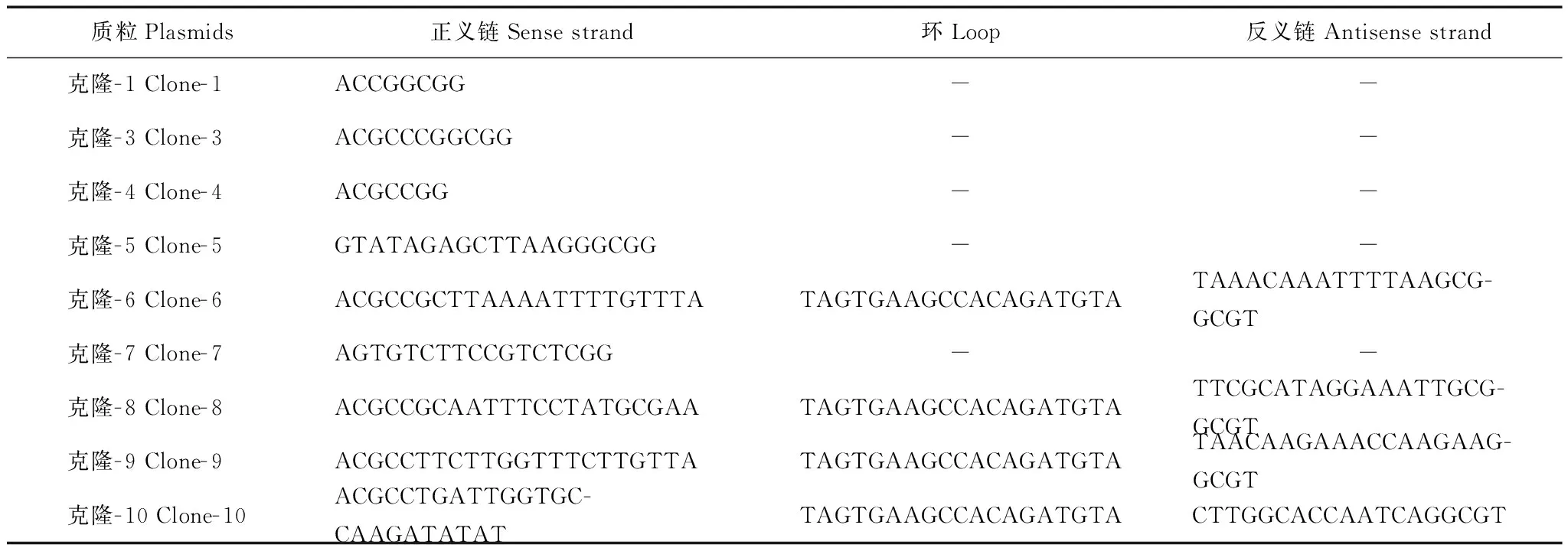
表 1 基于miRNA架构的shRNA随机文库质粒测序结果Table 1 Sequencing result of the plasmids from the miRNA context-based random shRNA library
-:无
-:none
本研究构建的新型大容量随机shRNA文库可通过置换其pol Ⅱ启动子用于组织特异性大规模RNAi筛选研究;也可将基于miRNA架构的shRNA表达框置于报告基因的3’端,这样可以同时检测报告基因的表达和shRNA的干扰表型;还可以多顺反子形式同时表达多个shRNA用于组合RNAi研究,并且不会干扰内源miRNA通路从而避免细胞毒性[1],为功能基因组的研究提供了有力的工具。
[1]Fellmann C,Hoffmann T,Sridhar V,et al.An optimized microRNA backbone for effective single-copy RNAi [J].Cell Reports,2013,5(6):1704- 1713.
[2]Stegmeier F,Hu G,Rickles RJ,et al.A lentiviral microRNA-based system for single-copy polymerase Ⅱ-regulated RNA interference in mammalian cells [J].Proc Natl Acad Sci USA,2005,102(37):13212- 13217.
[3]Yang G,Yuan L,Lu X,et al.A rational design of completely random shRNA library [J].Biochem Bioph Res Co,2013,430(3):987- 992.
[4]Wang Y,Wang YE,Cotticelli MG,et al.A random shRNA-encoding library for phenotypic selection and hit-optimization [J].PLoS One,2008,3(9):e3171.
[5]Grimson A,Farh KK,Johnston WK,et al.MicroRNA targeting specificity in mammals:determinants beyond seed pairing [J].Mol Cell,2007,27(1):91- 105.
[6]Lin X,Ruan X,Anderson MG,et al.siRNA-mediated off-target gene silencing triggered by a 7 nt complementation [J].Nucleic Acids Res,2005,33(14):4527- 4535.
[7]Chen M,Zhang L,Zhang HY,et al.A universal plasmid library encoding all permutations of small interfering RNA [J].Proc Natl Acad Sci USA,2005,102(7):2356- 2361.
[8]Bhinder B,Djaballah H.A simple method for analyzing actives in random RNAi screens:introducing the “H Score” for hit nomination & gene prioritization [J].Comb Chem High Throughput Screen,2012,15(9):686- 704.
Construction of A New Random Short Hairpin RNA Library Based on microRNA Context
GUO Qian,ZHU Ning,BU Mengmeng,GUO Sichao,YAN Xuejing,CHEN Qian,CHEN Meihong
State Key Laboratory of Medical Molecular Biology,Department of Biochemistry and Molecular Biology,Institute of Basic Medical Sciences,CAMS and PUMC,Beijing 100005,China
CHEN Meihong Tel:010- 69156410,E-mail:chenmeihong@hotmail.com
Objective To build an efficient random short hairpin RNA(shRNA)library.Methods shRNA expression vector was constructed with enhanced green fluorescent protein(EGFP)in the upstream of shRNA,driven by pol Ⅱ promoter(CMV).After the constructs were transfected into cells,the proteins were collected.The inhibition efficiency of shRNA was determined by Western blot and dual luciferase reporter system.After the shRNA expression vector was constructed with EGFP in the upstream of shRNA,driven by pol Ⅱ promoter(CMV),shRNA was further embedded into microRNA(miRNA)context.The constructs were transfected into cells,and then the inhibition efficiency of shRNA against target genes was evaluated by quantificational real-time polymerase chain reaction.According to the result of quantificational real-time polymerase chain reaction,a new random shRNA library was constructed based on miRNA context.Results shRNA downstream of a large transcript was transcripted efficiently by pol Ⅱ promoter(CMV).The efficiency of shRNA interference on target gene was improved when shRNA was embedded into miRNA context.Thus,we constructed a new random shRNA library sized 1.8×107based on miRNA context.Conclusion We successfully constructed a new large random shRNA library.
microRNA;short hairpin RNA;random library
518-524
shRNA:短发夹RNA;NOV:肾母细胞瘤过度表达基因;EGFP:增强绿色荧光蛋白;Mr:相对分子质量;Blank:未转染质粒的MG63细胞;1.转染pLenti6.3载体;2.转染pLenti6.3-EGFP;3.转染pLenti6.3-shNOV;4.转染pLenti6.3-EGFP-shNOV;与pLenti6.3比较,aP<0.01
shRNA:short hairpin RNA;NOV:nephroblastoma over-expressed gene;EGFP:enhanced green fluorescent protein;Mr:relative molecular mass;Blank:untransfected MG63 cell;1.cells transfected with pLenti6.3 vector;2.cells transfected with pLenti6.3-EGFP;3.cells transfected with pLenti6.3-shNOV;4.cells transfected with pLenti6.3-EGFP-shNOV;aP<0.01 compared with pLenti6.3
A.MG63细胞转染shRNA表达质粒后靶基因NOV的蛋白水平;B.HEK293T细胞转染shRNA表达质粒后靶基因Firefly荧光素酶的活性
A.protein level of NOV in MG63 cells transfected with shRNA expression plasmids;B.firefly luciferase activity in HEK293T cells transfected with shRNA expression plasmids
国家自然科学基金(31050008、31470067)Supported by the National Natural Sciences Foundation of China(31050008,31470067)
陈梅红 电话:010- 69156410,电子邮件:chenmeihong@hotmail.com
Q74
A
1000- 503X(2017)04- 0518- 07
10.3881/j.issn.1000- 503X.2017.04.010
2016- 04- 13)
猜你喜欢
湘潮(上半月)(2022年7期)2022-12-06
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
天津医科大学学报(2021年4期)2021-08-21
猪业科学(2021年3期)2021-05-21
江西农业学报(2021年4期)2021-04-20
中日友好医院学报(2021年1期)2021-04-14
广东蚕业(2021年1期)2021-03-18
幽默大师(2020年10期)2020-11-10
三农资讯半月报(2020年11期)2020-06-21
