
二代测序法用于先天性长QT综合征临床基因检测的假阴性分析
2017-09-03 10:31李新刘念白融冯莉阮燕菲马长生
中国循环杂志 2017年8期
李新,刘念,白融,冯莉,阮燕菲,马长生
二代测序法用于先天性长QT综合征临床基因检测的假阴性分析
李新,刘念,白融,冯莉,阮燕菲,马长生
目的:探讨二代测序法在先天性长QT综合征(LQTS) 临床基因检测中的假阴性问题。
方法:选取2个商业医学检验实验室(Lab1 和Lab2,HiSeq2000测序平台)、1个商业科研服务实验室(Lab3,Ion Torrnet测序平台)和1个学术机构实验室(Lab 4,HiSeq2000测序平台)产生的共28例样本数据(Lab1:6例;Lab2:8例;Lab3:8例;Lab4:6例),定量分析LQTS的三个主要致病基因KCNQ1、KCNH2和SCN5A外显子区域测序覆盖度以及可能漏检的致病变异数目。
结果:采用HiSeq2000测序平台的3个实验室(Lab1、Lab2和Lab4)中,三个致病基因外显子区域覆盖度>10倍的比例均高于98%,覆盖度>30倍的区域介于90%~95%。KCNQ1在两个商业医学检验实验室的14例样本中,低于10倍和30倍覆盖的外显子区域比例平均为3.63%和9.84%;低于10倍覆盖区域集中在第一外显子,平均包含约2%的已知致病或疑似致病变异。KCNH2在两个商业医学检验实验室14个样本中,低于10倍和30倍覆盖的区域分别为2.64%和15.76%,低覆盖区分布在多个外显子中。Lab1的数据中,KCNH2低于30倍覆盖区域最高达28.56%,其内包含已知致病或疑似致病变异113个(19.79%)。SCN5A的整体覆盖度最好,四个实验室的数据都不存在低于10倍覆盖的区域,其中两个商业医学检验实验室也不存在低于30倍覆盖的区域。
结论:当前的LQTS基因二代测序检测中,KCNQ1和KCNH2都存在一定程度的低覆盖区,因此普遍存在漏检致病变异的可能,假阴性问题值得高度重视。
QT延长综合征;高通量核苷酸测序;基因;假阴性反应
(Chinese Circulation Journal, 2017,32:771.)
先天性长QT综合征(LQTS)是一种遗传性心律失常。最近的数据表明LQTS发病率达1/2500~1/2000[1]。基因检测对于LQTS的诊断和治疗有重要意义,而对儿茶酚胺性敏感性多形性室性心动过速、Brugada综合征等心律失常基因检测目前仍无法指导治疗[2]。因此,与其他心律失常相比,基因检测对于LQTS的临床价值更为突出,可以说是心血管领域尤其是心律失常领域应用基因检测的典范。由于二代测序(NGS)技术高通量的优点,当前不仅在科研领域广泛应用,在我国临床基因检测领域也逐渐取代传统一代测序(Sanger测序)。然而,NGS在临床应用中仍缺乏统一的质控标准或共识,迫切需要大量的质量控制研究以促进相关标准的建立,从而更好的支持NGS在精准医学中的应用。
本文选取2个商业医学检验实验室、1个商业科研服务实验室和1个学术机构实验室应用不同NGS平台产生的数据,对LQTS的三个主要致病基因KCNQ1、KCNH2和SCN5A测序数据质量进行评估,分析NGS在目标基因覆盖以及变异位点漏检方面的特点,探讨NGS用于临床检测LQTS的假阴性问题。
1 资料与方法
1.1 研究对象
选取欧洲离子通道和心肌病遗传检测专家共识中推荐的LQT1-3的3个基因(KCNQ1、KCNH2和SCN5A)为研究对象[2]。分析这3个基因的测序覆盖度和测序未覆盖区(“缺口”)中包含的“致病”或“可能致病”突变个数,从而评估当前NGS用于LQTS遗传诊断带来的假阴性问题。
1.2 数据来源
NGS数据来自2个商业医学检验实验室(Lab1、Lab2,拥有临床细胞分子遗传学检验资格)、1个商业科研服务实验室(Lab3)和1个学术机构实验室(Lab4)。测序样品均为外周血提取的基因组DNA,测序目标基因都包含KCNQ1、KCNH2和SCN5A。Lab1提供了6例遗传性心律失常基因检测患者的数据,方法为Agilent靶向捕获探针对213个心血管相关基因进行捕获,用HiSeq2000平台(Illumina公司)生成测序数据。Lab2提供了8例遗传性心血管病基因检测患者的数据,其中4例采用Roche探针捕获445个基因的心血管检测包,4例采用Agilent探针捕获6110个基因的综合检测包,测序平台也为HiSeq2000。Lab3提供了8例科研用途的心律失常患者数据,目标区域包括50个基因,测序平台为Ion Torrent。Lab4提供了6例科研用途的健康人外显子组测序数据(Agilent 50Mb或70Mb试剂盒),测序平台也为HiSeq2000。需要说明的是,这些数据并不能用于比较这4个实验室或其所用平台的测序质量优劣,我们主要目的是从这些数据中探讨假阴性问题的普遍性。
1.3 分析方法
对于NGS原始数据,首先进行基因组比对,比对前使用cutadapt软件去除接头和低质量序列,仅保留连续Q值>20的碱基进行比对。每测一个碱基有一个相应的质量值(Quality,Q),用来衡量测序准确度。碱基的Q值为20相当于错误率为1%。使用BWA(v0.7.15)软件和人的基因组参考序列(hg19)进行比对,采用Picard(v2.5.0)软件去除聚合酶链式反应(PCR)重复引入的测序片段(reads),使用Samtools(v1.3.1)软件将sam文件转换成bam文件并按染色体排序和建立索引。
测序覆盖度分析采用BEDTools软件包中的“Coverage analysis for targeted DNA capture”功能对目标区域reads覆盖度进行统计(http://bedtools. readthedocs.io/en/latest/,美国犹他大学)。BEDTools是用于各种基因组特征比较及注释的工具[3]。目标基因编码区参考序列为KCNQ1(NM_000218.2)、KCNH2(NM_000238.3)和SCN5A(NM_198056.2),编码区长度分别为2031 bp、3480 bp和6051 bp,累计长度11562 bp。软件计算出每个碱基位置的测序覆盖度,连续5个碱基覆盖度低于10倍或30倍,则认为此区域为<10倍或<30倍。>10倍覆盖的比例是绝大多数研究中都会报道的NGS数据指标;而>30倍覆盖通常被认为是高可信度的标准,在近期发布的临床基因检测研究中也有应用实例[4,5]。逐个样本记录这3个基因上的低覆盖区域碱基数,再按实验室分组统计覆盖度的平均数±标准差。
1.4 临床变异位点参考数据库
参考数据库为美国国家生物技术信息中心(NCBI)的ClinVar数据库[6,7](http://www.ncbi.nlm. nih.gov/clinvar/)。对于每个样本数据中的低覆盖区域,记录其在数据库中相应区域内包含的致病变异和疑似致病变异个数。
2 结果
2.1 4组来自不同实验室测序数据的覆盖度评估
用HiSeq 2000测序平台的3个实验室KCNQ1、KCNH2和SCN5A的覆盖度都在98%以上(至少10倍覆盖),采用外显子组测序的Lab4,<30倍覆盖的比例明显高于两个临床医学检验实验室。而采用Ion Torrent的Lab3,数据覆盖度略低于其他实验室(图1),这与Lab3提供数据的实验设计有关,并不意味着Ion Torrent平台的覆盖度不如其它平台。实际上,所有实验室的>10倍和>30倍覆盖区域所占比例差异无统计学意义(P>0.05)。
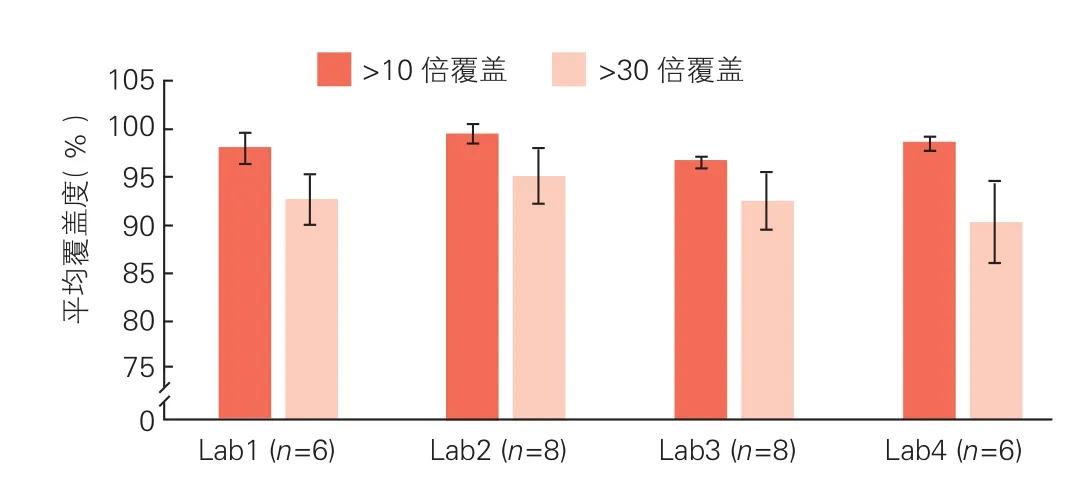
图1 KCNQ1、KCNH2和SCN5A三个基因的总体测序覆盖情况
2.2 低覆盖区域比例及包括的变异数
Clinvar数据库(2016-08-10)收录的KCNQ1、KCNH2和SCN5A中致病及可能致病的(Pathogenic / Likely pathogenic)变异个数分别为:KCNQ1中386个,KCNH2中571个,SCN5A 中442个。
KCNQ1:28例样本中有5例样本(Lab2的4个,Lab3的1个)此基因被完全覆盖(>10倍),其余23例样本低覆盖度区域集中在第1个外显子(表1)。以Lab1中此基因覆盖最差的1号样本为例,编码区开头的178 bp覆盖度低于10倍(8.7%),包括7个ClinVar数据库收录的致病或疑似致病变异,占此基因全部386个的1.8%;<30倍区域287 bp,包含10个致病或疑似致病变异。Lab2中此基因覆盖较差的7号样本有192个碱基覆盖度低于10倍,其中包括8个ClinVar数据库收录的致病或疑似致病变异,占此基因所有386个的2%;<30倍区域319 bp,包含15个致病或疑似致病变异。两个临床检验实验室14个样本(表1),KCNQ1中<10倍的外显子区域比例平均(3.63±0.37)%,<30倍区域比例平均(9.51±0.41)%。

表1 KCNQ1在三个HiSeq平台实验室的测序覆盖情况
KCNH2:4个实验室中Lab3数据在KCNH2覆盖度最差,在4个外显子上存在<10倍区域,因其所用测序平台不同于其它三个实验室,数据未列在表2中。两个临床检测实验室中,Lab2的覆盖情况优于Lab1,8个样本中有5个样本此基因存在<10倍区域。而Lab1的6个样本均有存在<10倍区域,与采用外显子捕获测序的Lab4很相似,低覆盖区域主要集中在4号和12号外显子(表2)。两个临床检验实验室14个样本,KCNH2中<10倍和<30倍覆盖度的外显子区域比例平均值分别为2.64%和15.76%。
以Lab1中覆盖较差的1号样本为例,覆盖度<10倍区域累计236 bp,占此基因编码区的6.78%。这些低覆盖区域中包含31个ClinVar数据库收录的致病或疑似致病变异,占所有571个变异的5%。此样本<30倍覆盖区域更是高达28.56%(表2),其中包括致病或疑似致病变异113个(19.79%)。

表2 KCNH2在三个HiSeq平台实验室的测序覆盖情况
SCN5A:SCN5A是三个基因中覆盖度最好的。4个实验室所有样本SCN5A各外显子测序覆盖度均>10倍,两个临床检测实验室的数据覆盖度均>30倍。采用外显子组测序的学术机构实验室Lab4 此基因<30倍覆盖的平均比例为2.9%。
3 讨论
我们通过对4个实验室28例样本NGS数据的分析,首次展示了国内当前采用NGS检测LQTS基因变异将不可避免地出现低覆盖区域,从而引起致病突变漏检(假阴性)的可能。本研究观察到两个临床检验实验室的数据SCN5A测序覆盖情况良好;而KCNQ1和KCNH2<10倍覆盖区域平均值分别为3.38%和2.64%,在不同样本间差异较大(0%~11.6%)。美国著名基因检测公司Familion以1例健康个体的Ion Torrent NGS数据为例,指出仅用NGS在KCNQ1、KCNH2、SCN5A和RYR2四个重要心律失常基因上存在不容忽视的低覆盖区,比如KCNH2<40倍覆盖的区域达34%[8]。
NGS覆盖度受测序数据量、目标基因序列GC含量、目标区域捕获效率、检测包(Panel)中基因之间序列的相似性程度等影响,不同批次送检的同一Panel的测序结果之间也会有一定程度的差异。因此,Familion指出为提高临床基因检测结果的准确性,应联合采用经典Sanger测序和NGS。一些国外著名临床机构对遗传性心律的基因检测也采用Sanger测序为代表的其它方法对NGS覆盖不满意的区域进行补充,基本不允许存在“缺口”,但对需要补充Sanger测序或其它方法的区域并没有统一标准(Dr. Ackerman 私人通讯2015)。对关键基因做到完全覆盖,有利于提高阳性检出率。我国最大的LQTS注册队列研究,通过心电图预测指导的Sanger测序法仅对这3个基因进行顺序检测,在230例患者中突变检出率达81%(186/230)[9]。
理论上,用于临床检测的NGS质量控制应比科研目的的检测质量控制更加严格,但实际上,国内很多临床基因检测公司对于影响测序质量的很多环节的处理与科研检测无异,质量控制还有待改善。这种情况与国内临床基因检测领域尚无可遵循的指南或标准也有关系。2015-12欧洲率先发表NGS临床诊断应用指南[10],其第一条就指出“阻止NGS过早用于诊断的原因只能是其质量差,未通过验证就进入临床诊断对患者而言具有威胁性,同时也是不可接受的”。值得注意的是,最近一些临床基因检测公司对于临床检测样本和科研样本已区别对待,对临床检测样本会适当提高测序数据量(测序深度),这可在很大程度上缩小低覆盖区域的比例。但仅靠提高测序数据量还不够,如上所述,测序的质量还受很多其它因素的影响,如建库质量、捕获效率以及目标基因本身的序列特征等影响。对于特定基因,比如本文关注的3个基因在同一实验室的同一检测包中覆盖情况都有不同。
欧洲NGS临床诊断应用指南提出了NGS诊断检测的“评价系统”,将NGS诊断检验分为三类:A类:实验室可保证编码区及侧翼区99%以上的测序可靠度,通过Sanger 测序或其它方法补充NGS漏洞区域的序列。B类:实验室明确指出哪些区域的NGS测序可靠度在99%以上,选择性的对某些NGS漏洞区域进行Sanger 测序或其它方法补充。C类:实验室测序仅依赖NGS,无Sanger或其它测序补充。目前国内的NGS临床诊断商业公司,绝大多数都为C类,在基因筛查阶段只依赖NGS,只是在检测到个别相关性强的位点时,才会进行此位点的Sanger测序验证。因此,目前国内NGS临床检测的假阴性的问题是比较普遍且严重的。心血管方面尚缺乏公开报道的数据,而在眼科相关基因检测中,我国学者通过对179个视网膜色素变性家系先证者进行NGS,50人未能检测到相关变异。对这50人进一步用Sanger 测序法重新检测原来NGS已包括的ORF15基因,又在7例患者中发现了致病突变,而这7例患者的致病突变都位于NGS的低覆盖区域(<10倍)[11]。
本文的局限性在于,仅对4个实验室测序数据比对上的部分进行了覆盖度分析,未能具体分析低覆盖区域产生的技术原因。但这4个实验室的数据普遍表明了仅用二代测序进行LQTS临床基因检测存在不容忽视的假阴性可能,临床医生或诊断实验室应谨慎下结论。
综上,假阴性率是临床基因检测中的重要质量控制问题之一。对于遗传性心律失常的临床基因检测,我们呼吁对于指南或共识中[12]推荐的基因应在NGS外采补充Sanger测序,以尽量减少假阴性,提高心律失常NGS临床检测准确率。我们也希望我国能尽快出台NGS临床应用相关的指南或标准,引导NGS在精准医疗事业中发挥更大价值。
[1] Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation, 2009, 120: 1761-1767.
[2] Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm, 2011, 8: 1308-1339.
[3] Quinlan AR, Hall IM. Bedtools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 2010, 26: 841-842.
[4] Manase D, D'Alessandro LC, Manickaraj AK, et al. High throughput exome coverage of clinically relevant cardiac genes. BMC Med Genomics, 2014, 7: 67.
[5] Megahed H, Nicouleau M, Barcia G, et al. Utility of whole exome sequencing for the early diagnosis of pediatric-onset cerebellar atrophy associated with developmental delay in an inbred population. Orphanet J Rare Dis, 2016, 11: 57.
[6] Rehm HL, Berg JS, Brooks LD, et al. ClinGen--the Clinical Genome Resource. N Engl J Med, 2015, 372: 2235-2242.
[7] Landrum MJ, Lee JM, Riley GR, et al. Clinvar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2014, 42: D980-985.
[8] Callis TE, Hutchinson S, Lu H, et al. Complementary use of conventional and nextgeneration sequencing methods is critical for accurate mutation detection in multi-gene arrhythmia panels. Heart Rhythm, 2015, 12(Suppl): S97-S154.
[9] Gao Y, Liu W, Li C, et al. Common genotypes of long QT syndrome in China and the role of ECG prediction. Cardiology, 2016, 133: 73-78.
[10] Matthijs G, Souche E, Alders M, et al. Guidelines for diagnostic nextgeneration sequencing. Eur J Hum Genet, 2016, 24: 2-5.
[11] Huang XF, Wu J, Lv JN, et al. Identification of false-negative mutations missed by next-generation sequencing in retinitis pigmentosa patients: A complementary approach to clinical genetic diagnostic testing. Genet Med, 2015, 17: 307-311.
[12] 中华心血管病杂志编辑委员会心律失常询证工作组. 遗传性原发性心律失常综合征诊断与治疗中国专家共识. 中华心血管病杂志, 2015, 43: 5-20.
False-negative Possibility in Genetic Test of Congenital Long QT Syndrome by Next-generation Sequencing
LI Xin, LIU Nian, BAI Rong, FENG Li, RUAN Yan-fei, MA Chang-sheng.
Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, National Clinical Research Center for
Cardiovascular Diseases, Beijing (100029), China
MA Chang-sheng, Email: chshma@vip.sina.com
Objective: To explore the false-negative possibility in genetic test of congenital long QT syndrome (LQTS) by nextgeneration sequencing (NGS).
Methods: A total of 28 genomic DNA samples were collected from 4 laboratories including 2 commercial medical laboratories using HiSeq2000 platform as Lab1, n=6 and Lab2, n=8; 1 commercial research service laboratory using Iontorrent platform as Lab3, n=8 and 1 academic laboratory using HiSeq2000 platform as Lab 4, n=6. Sequencing coverage in the exons of protein-coding region in 3 main LQTS pathogenic genes as KCNQ1, KCNH2, SCN5A and possible pathogenic variants were quantitatively analyzed.
Results: In Lab1, Lab 2 and Lab 4 with HiSeq2000 platform, above 98% protein coding regions in 3 pathogenic genes were covered with>10-fold reads and 90%-95% were covered with>30-fold reads. In 2 commercial medical laboratories, 3.63% and 9.84% protein coding regions of KCNQ1 gene in 14 samples were covered with<10-fold reads and with<30-fold reads; lower than 10-fold covering region was focused in the 1stexon including about 2% known or likely pathogenic variants. In 2 commercial medical laboratories, 2.64% and 15.76% protein coding regions of KCNH2 gene in 14 samples were covered with<10-fold reads and with<30-fold reads; low covering region was located in multiple exons. For the data from Lab 1, ashigh as 28.56% protein coding regions of KCNH2 gene were covered with<30-fold reads including 113 (19.79%) known or likely pathogenic variants. SCN5A gene had the best coverage of protein coding region, with no<10-fold reads in all 4 Labs and no<30-fold reads in 2 commercial medical laboratories.
Conclusion: Currently, NGS has low coverage region in both KCNQ1 and KCNH2 genes, pathogenic variants could be missed and false-negative possibility should be highly alert.
Long QT syndrome; High-throughput nucleotide sequencing; Gens; False Negative Reactions
book=771,ebook=47
2016-11-03)
(编辑:漆利萍)
国家自然科学基金(81500246,81470465);北京市自然科学基金(7161003);北京市医管局临床医学发展专项(ZYLX201302)
100029 北京市,首都医科大学附属北京安贞医院 心内科, 国家心血管病临床医学研究中心
李新 副研究员 博士 主要从事心血管病遗传与基因组学研究 Email: leexin9907@126.com 通讯作者: 马长生 Email: chshma@vip.sina.com
R54
A
1000-3614(2017)08-0771-05
10.3969/j.issn.1000-3614.2017.08.010
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
科学技术创新(2022年30期)2022-10-21
农业与技术(2021年23期)2021-12-14
黑龙江水利科技(2020年8期)2021-01-21
中国生殖健康(2020年4期)2021-01-18
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中央民族大学学报(自然科学版)(2018年2期)2018-11-09
中国生殖健康(2018年4期)2018-11-06
百科知识(2015年18期)2015-09-10
