
婴儿型脊肌萎缩症的神经电生理特点
2017-08-07 04:26吴雪燕李红勤
临床神经病学杂志 2017年3期
吴雪燕,李红勤
·学术交流·
婴儿型脊肌萎缩症的神经电生理特点
吴雪燕,李红勤
目的 探讨婴儿型脊肌萎缩症(SMA)的神经电生理特征。方法 采用肌电/诱发电位仪对27例SAM患儿进行神经传导速度测定,每例进行至少6块肌肉的EMG检查,分析检查结果。结果 婴儿型SMA正中神经复合肌肉动作电位(CMAP)波幅显著下降,尺神经与腓总神经的CMAP波幅明显下降,伴有尺神经运动传导速度的轻度减慢;所记录感觉神经传导未见明显异常。EMG提示神经源性损伤。结论 婴儿型SMA典型临床症状为进行性加重的对称性肌肉无力和萎缩,特异性的神经电生理表现为本病的诊断提供重要的依据。
婴儿型脊肌萎缩症;EMG;神经运动传导
脊肌萎缩症 (SMA) 是一种以脊髓前角细胞变性为主要病理改变的常染色体隐性遗传病,居致死性常染色体隐性遗传病的第二位[1]。在18岁以下儿童诊断明确的神经肌肉疾病之中,SMA的发病率列为第三位[2]。SMA的主要特征为下运动神经元变性导致的进行性肌肉无力以及肌萎缩,肌张力减低。我国在SMA方面尚无流行病学的完整资料。近年来随着遗传学的发展,婴儿型SMA的检出率有所提高。本研究通过对27例临床确诊的婴儿型SMA患儿进行EMG检查,探讨其神经电生理特点。
1 对象与方法
1.1 对象 选择2011年1月~2015年11月在我院确诊的SMA患者27例。男16例,女11例;年龄3.5个月~24个月,平均12.2个月;发病年龄15 d~8个月,平均发病年龄为3.1个月。病程5月~14月,平均9.4个月
1.2 方法
1.2.1 神经传导检测 应用KEYPOINTEMG/诱发电位仪进行外周神经运动及感觉传导检测和EMG检查。运动神经传导速度(MNCV)以及肌肉复合动作电位(CMAP)波幅和同年龄正常值的低限比较[3]:下降<30%视为轻度下降,下降>50%视为显著下降;远端潜伏期和同年龄正常值的高限比较:延长<30%视为轻度延长,延长>50%视为显著延长。感觉神经传导速度(SCV)以及波幅和同年龄正常值的低限比较[3]:下降<30%视为轻度下降,下降>50%视为显著下降。行周围神经运动传导检查时,采用马鞍桥刺激电极,以时限为0.2 ms的方波脉冲给予超强刺激,在腕部和肘部体表分别刺激正中神经与尺神经,在踝部和腘窝体表分别刺激胫神经与腓总神经,用表面电极分别在对应侧的拇短展肌、小指展肌、踇展肌以及趾短伸肌的皮肤表面记录CMAP波形,测定所检测神经两个刺激点的动作电位潜伏期以及两点间距离,计算该神经段的MNCV,分析所检神经的远端潜伏期,CMAP波幅,以及MNCV[3]。行周围神经感觉传导检查时,同样采用马鞍桥刺激电极,用顺向法分别在中指和小指刺激正中神经与尺神经,表面电极分别在对应的腕部记录感觉神经动作电位;用逆向法在小腿外侧缘刺激腓浅神经,表面电极位于对应足背记录。测定所检测神经刺激点到记录电极距离,计算出该神经段的SCV,分析所检感觉神经电位的波幅以及SCV[3]。
1.2.2 EMG检查 行EMG检查时,选择四肢由不同神经支配的至少6块以上的肌肉。局部皮肤消毒,将同轴同心针电极快速刺入肌肉。患儿均为婴幼儿,不能完全配合动作,故各状态均为不自主动作下记录。在不自主放松时进行多部位探测,观察波形,监听声音,观察是否有纤颤电位,正相波,肌强直电位等自发电位;然后在肌肉不自主轻收缩时观察并且采集运动单位电位(MUP)的波形,测定平均时限;在不自主重收缩时观察募集相。各运动感觉神经传导数值正常值参见文献[3-4]。
2 结 果
2.1 神经传导检测
2.1.1 运动神经传导检测 见表1。共检测四肢神经84条,有2条尺神经、1条正中神经、7条腓总神经无诱发波幅。引出的神经中,有17.57%(13/74例)的CMAP波幅下降至500 μV以下,其中,22例正中神经的CMAP波幅显著下降,平均下降51.56%,18例尺神经、29例腓总神经的CMAP波幅分别平均下降33.5%和47%;18例尺神经的MNCV平均减慢2.73%,其他神经MNCV均未出现减慢;远端潜伏期未见明显延长。
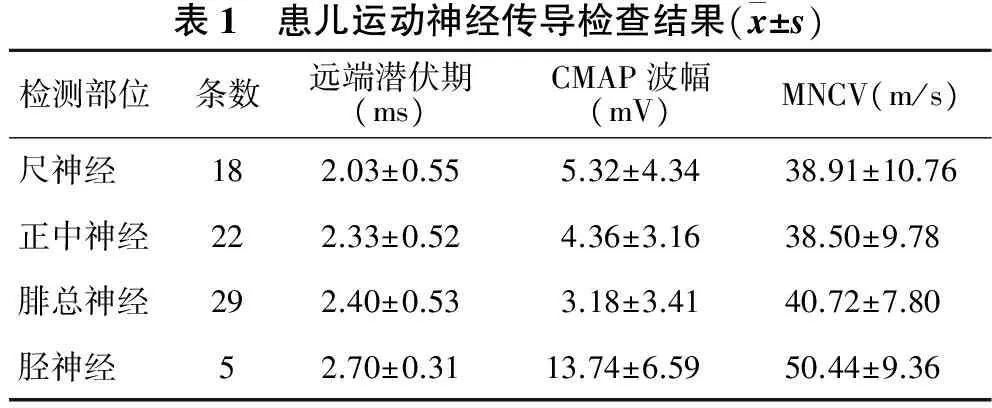
表1 患儿运动神经传导检查结果(x±s)检测部位条数远端潜伏期(ms)CMAP波幅(mV)MNCV(m/s)尺神经182.03±0.555.32±4.3438.91±10.76正中神经222.33±0.524.36±3.1638.50±9.78腓总神经292.40±0.533.18±3.4140.72±7.80胫神经52.70±0.3113.74±6.5950.44±9.36
2.1.2 感觉神经传导检测 见表2。共检测四肢神经68条,所检神经中,尺神经、正中神经和腓浅神经的感觉神经电位波幅均稍有减低,分别平均减低8.04%、4.26%和9.86%;SCV均在正常范围。
2.2 针电极EMG结果 见表3。27例患儿共检测肌肉238块,其中有19块肌肉不能完全放松,不能观察其静息状态时是否有自发电活动(包括胸锁乳突肌、腹直肌)。余肌肉静息时纤颤电位、正相波出现率为100%(219/219),其中11块肌肉纤颤电位及正相波数量较多。所检肌肉中可见较多肌肉MUP时限增宽,其中上肢肌31.25%(30/96),下肢肌47.87%(45/94)。部分肌肉可见明显多相电位出现。27例患儿均表现为四肢活动减少,其不自主最大用力收缩时MUP募集明显减少,呈现为单纯相或者单纯混合相,有29块肌肉仅出现少量运动单位。
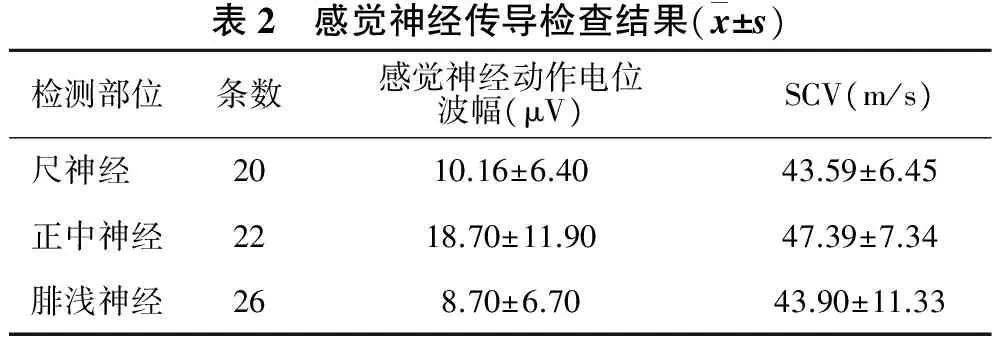
表2 感觉神经传导检查结果(x±s)检测部位条数感觉神经动作电位波幅(μV)SCV(m/s)尺神经2010.16±6.4043.59±6.45正中神经2218.70±11.9047.39±7.34腓浅神经268.70±6.7043.90±11.33

表3 EMG检测结果(例,%)肌肉总数(块)自发电位MUP增宽多相电位第一背侧骨间肌3333(100)7(21.21)11(33.33)拇短展肌1010(100)3(30)2(20)伸指总肌2424(100)7(29.17)11(45.83)桡侧腕屈肌99(100)4(44.44)3(33.33)三角肌1111(100)5(45.45)5(45.45)肱二头肌1414(100)4(28.57)6(42.86)踇长伸肌77(100)3(42.86)3(42.86)胫前肌4545(100)18(40)24(53.33)腓肠肌内侧头3737(100)14(37.84)4(10.81)股内肌2020(100)8(40)7(35)股直肌55(100)2(50)2(40)冈下肌22(100)0(0)1(50)腹直肌22(100)2(100)1(50)
3 讨 论
婴儿型SMA是最早由Werdnig(1891年)和Hoffmann(1893年)报道,故又名Werdnig-Hoffmann病[5]。我国在此疾病方面尚无全面完整的流行病学资料,而至2001年为止,中国南方此病的发病率在活产婴儿当中为1/53 000[6]。1995年以前,SMA的诊断主要依靠病史、临床表现、EMG及肌肉活检,但是肌肉活检创伤性较大,特异性欠佳,在临床推广困难。近年来随着医学的发展,SMA的基因学研究已取得了突破性的进展。研究[7]发现,约 98.6%的患者有 SMN 基因第 7、第 8 外显子纯合缺失或突变,已公认 SMA 的发病主要是 SMNt 基因缺失引起,且缺失的程度和范围与临床表型的严重程度密切相关[8-9]。因此对SMN 基因检测已作为诊断 SMA的主要判断依据。然而在本地区,SMN基因检测尚未普及,主要诊断手段仍依靠临床病史及EMG、肌肉活检检测,而EMG检测证据有助于建议患儿家属针对性的进行SMN基因检测,减少其就医成本,提高SMA早期诊断率,有助于达成早期诊断,尽可能早的进行临床干预。本研究中的27例入组患儿均为临床病史疑似,并于无锡市人民医院行EMG检查疑诊后至外院行基因检测确诊为婴儿型SMA的患者。
本研究中病例特点包括:(1)入组患儿均在8个月之内发病;(2)主要临床表现为肢体受累,双下肢活动减少为突出表现,少数患儿呈现蛙状腿;(3)体检可发现四肢肌力、肌张力均有所下降,以下肢为著,腱反射减弱或消失;(4)神经传导及EMG检查提示神经源性改变,外周神经CMAP波幅显著下降。
神经传导检测和针电极EMG检查是诊断SMA的重要依据。本研究中的27例患儿均接受了神经传导以及EMG检查,共检查四肢周围神经152条。结果显示,正中神经的CMAP波幅显著下降(下降>50%),尺神经、腓总神经的CMAP波幅均出现明显下降,而所检神经中仅见尺神经MNCV出现轻度减慢,余神经未见明显减慢,与沈瑛等[10]的研究结果相印证。SMA的基本病理变化是脊髓前角细胞变性,导致运动神经元损伤,轴突破坏,因此,在神经传导检测中,表现为所有刺激部位成功诱发的CMAP波幅均降低,伴有MNCV的相对正常或者轻度减慢,而对SCV以及波幅影响相对较小,这与朱敏等[11]观察结果相近。亦有文献[12]报道婴儿型SMA患儿出现感觉神经传导速度显著减慢,可能和本病并发的后根、脊神经节、上行纤维以及丘脑等损害有关,但是此过程较运动神经元病变发展更缓慢,并且可能伴随其他的病变[13]。
婴儿型SMA异常表现常为多样性,可同时出现广泛的失神经现象以及慢性神经再生现象,异常部位常为延髓,颈、胸、腰段支配的区域。即使是在病变的早期,对临床上尚未受累阶段支配的肌肉也可以看到正在进行的轴索变性,表现为宽时限高波幅的巨大再生电位,失神经电位主要表现为纤颤电位和正相波。本研究EMG检查显示,所有病例在静息状态下均出现纤颤电位与正相波(出现率100%),呈现失神经改变,轻收缩时部分MUP时限增宽(其中上肢肌占31.25%,下肢肌占47.87%),部分肌肉出现多相电位,均符合脊髓前角细胞受损的诊断,这与刘长云等[14]观察结果相近。而在不自主最大用力收缩时募集相呈现单纯相或者单纯混合相,并且有部分肌肉仅出现少量运动单位。但因为患儿基本不会自主用力,故此项结果仅为参考。
婴儿型SMA具有典型的临床与神经电生理表现,发病时间早,病情进展快,四肢肌力下降明显,神经传导检查提示CMAP波幅显著下降,EMG检查提示神经源性损伤。因此,对于临床表现为进行性对称性肢体肌力下降的新生儿以及婴儿,应该高度警惕本病,及时作出诊断与鉴别诊断。神经电生理检查可以用于鉴别神经源性和肌源性损伤,可为本病的诊断提供可靠的依据[15],神经电生理检查结果也能够为遗传学诊断方法的选择提供参考。
总之,婴儿型SMA典型的临床表现为进行性加重的对称性肌肉无力和萎缩,特异性的神经电生理表现为本病的诊断提供重要的依据,是本病早期诊断与鉴别诊断的重要组成部分。
[1]Munsat TL, Davies KE. International SMA consortium meeting [J]. Neuromuscul Disord, 1992, 2: 423.
[2]Younger DS, Iannaccone ST, ed. Motor Disorders[M]. Phialdelphia: Lippincott Willams-Wilkins, 2005: 349~350.
[3]崔丽英. 简明肌电图学手册[M] . 北京: 科学出版社, 2006: 183~211.
[4]卢祖能, 曾庆杏, 李承晏. 实用肌电图学[M] . 北京: 人民卫生出版社, 2000: 309~326.
[5]Cifuentes DC, Frugier T, Melki J. Spinal muscular atrophy [J]. Semin Pediatr Neurol, 2002, 9: 145.
[6]Chung B, Wong V, Ip P. Prevalence of neuromuscular diseases in Chinese children: a study in southern China [J]. J Child Neurol, 2003, 18: 217.
[7]Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene[J]. Cell, 1995, 80: 155.
[8]Le TT, Pham LT, Butchbach ME, et al. SMN Delta7, the major product of the centromeric survival motor neuron(SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN[J]. Hum Mol Genet, 2005, 14: 845.
[9]Frugier T, Nicole S, Cifuentes-Diaz C, et al. The molecular bases of spinal muscular atrophy[J]. Curr Opin Genet Dev, 2002, 12: 294.
[10]沈瑛, 张静敏, 周敏杰,等. 13例婴儿型脊髓性肌萎缩症临床及电生理分析[J]. 上海交通大学学报(医学版), 2010, 30: 147.
[11]朱敏, 张跃, 汤健,等. 婴儿型脊髓性肌萎缩症的临床及电生理特点[J]. 临床神经病学杂志, 2011, 24: 209.
[12]王学禹, 王念亮, 孙文秀. 脊肌萎缩症35例[J]. 实用儿科临床杂志, 2000, 15: 32.
[13]Cheliout HF, Barois A, Urtizberea A, et al. Evoked potentials in spinal muscular atrophy [J].J Child Neurol, 2003, 18: 383.
[14]刘长云, 王永芹, 季加芬,等. 儿童脊肌萎缩症23例临床特点及遗传学分析[J]. 临床儿科杂志, 2007, 25: 88.
[15]程敏, 李梅, 张琴. 儿童型脊髓性肌萎缩的临床和电生理分析[J]. 第三军医大学学报, 2009, 31: 646.
Diagnostic value of electrophysiological examinations in infantile spinal muscular atrophy
WUXue-yan,LIHong-qin.
DepartmentofElectroneurophysiology,WuxiPeople’sHospitalAffiliatedtoNanjingMedicalUniversity,Wuxi214000,China
Objective To explore electrophysiological features of infantile spinal muscular atrophy (SMA), and to evaluate diagnostic value of electrophysiological examinations in patients with SMA.Methods Tweenty-seven SMA children from Jan 2011 to Nov 2015 diagnosed in Wuxi People’s Hospital were enralled. All patients had neurogenic changes by EMG and finally confirmed by genetic test as SMA. Basic clinical data were retrospectively analyzed. Nerve conduction velocity was tested in each patient; While EMG was performed in no less than six muscles in each patient. Results All these patients were characterized by progressive flaccid paralysis in limbs. In all cases, amplitude of CMAP in median nerve was significantly decreased, apparent decreased in ulnar nerve and peroneal nerve; while mild decrease of MCV was appeared in ulnar nerve. Nothing abnormal were detected in sensory nerve conduction. EMG demonstrated neurogenic damage. Conclusion Typical clinical symptoms of infantile SMA are progressive symmetrical loss of muscle control and atrophy in limbs, and specificity electrophysiological representation provides very important basis for the diagnosis.
infantile spinal muscular atrophy;EMG;motor nerve conduction
214000 无锡市人民医院神经电生理室
李红勤
R746.4
A
1004-1648(2017)03-0221-03
2016-10-06
2016-10-29)
猜你喜欢
癫痫与神经电生理学杂志(2022年6期)2022-02-10
小学科学(学生版)(2019年10期)2019-11-16
中国外汇(2019年23期)2019-05-25
价值工程(2018年25期)2018-09-26
中国环境监察(2017年5期)2017-10-23
现代电生理学杂志(2016年1期)2016-07-10
西南交通大学学报(2016年4期)2016-06-15
华北电力大学学报(自然科学版)(2016年3期)2016-04-25
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国康复理论与实践(2015年7期)2015-05-09
