
配合物的结构和性质∗
2017-08-03 08:09马磊殷耀鹏丁晓彬董晨钟
物理学报 2017年6期
马磊 殷耀鹏 丁晓彬 董晨钟
(西北师范大学物理与电子工程学院,甘肃省原子分子物理与功能材料重点实验室,兰州 730070)
(2016年10月13日收到;2016年12月8日收到修改稿)
马磊 殷耀鹏 丁晓彬 董晨钟†
(西北师范大学物理与电子工程学院,甘肃省原子分子物理与功能材料重点实验室,兰州 730070)
(2016年10月13日收到;2016年12月8日收到修改稿)
利用密度泛函理论的B3LYP杂化方法,在相对论有效芯势模型下,使用Gaussian 03程序对IV价镎离子(Np4+)与硝酸根离子形成的几种配合物(n=1—6,q=−2—+3)的几何结构进行了优化,给出了其结构参数及性质.研究发现:Np4+与NO−3在结合过程中均以双齿模式结合,且Np4+与结合形成的配合物的Np—N键及Np—O键的键长最短,但Np4+与结合形成的Np(NO3)4配合物的结合能最大、结合最稳定.最后,进一步计算了Np(NO3)4配合物的红外光谱,通过与已有的实验数据对比的一致性,确认了本文计算结果的可靠性.
,密度泛函理论,结合能,红外光谱分析
1 引 言
随着社会和科学技术的发展,核能的使用越来越受到重视.但是,随之而来的是大量核废料处理问题也变得日益尖锐.目前研发的处理核废料主流方法是普雷克斯(PUREX)流程[1].在PUREX流程中,将核燃料在释放能量过程中所产生的裂变产物和逐渐积累起来的超铀元素,即乏燃料,溶解到硝酸之中,通过加入萃取剂以萃取方式将乏燃料中有可利用价值的铀(U)和钚(Pu)进行分离与回收.但是,分离与回收过程中会受其他超铀元素的影响,如镎(Np)元素[2,3].所以,分离Np元素对每个分离与回收过程至关重要.另一面,在溶解乏燃料的硝酸盐溶液中镎离子以IV,V和VI价态存在.其中,Np(V)在硝酸溶液中最稳定,但是在受硝酸浓度与氧化还原等条件的影响下,Np(V)部分转化为Np(IV)和Np(VI)[1].因此,在转化过程中Np(IV)和Np(VI)与NO−3所形成的镎的硝酸盐的结构及其性质的进一步研究显得尤为重要.
在过去的一段时间内,人们对Np(IV,V,VI)在硝酸中所形成的配合物已进行了一些研究.例如,Laidler[4]对Np(IV,V,VI)的硝酸盐在不同的环境中的磁性、红外光谱和X射线进行了实验测量.Skilling和Nazaroff用噻吩甲酰三氟丙酮萃取法研究了Np4+和的络合作用,得到了硝酸镎(IV)的水溶液的吸收光谱,并且算出硝酸根络合物的逐级稳定常数和总稳定常数的对数值[5].Yin等[6]对镎酰离子与结合形成的配合物结构的稳定性进行了详细研究,研究表明与结合得最强,分子最稳定.此外,也有研究采用密度泛函理论方法对与N,N,N′, N′-四甲基-3-乙二酸-戊二酰胺(TMOGA)[7]以及BTBP(bis-triazinyl bipyridine)类[8]配体的相互作用进行了表征.尽管对Np(IV,V,VI)及进行了一些研究,但是这些研究主要针对实验所获得的结论,理论计算较少且研究比较分散.因此,系统地获得Np4+及其与NO−3所形成的化合物的结构和性质显得更加重要.在本文中,利用B3LYP杂化密度泛函理论方法对Np4+与NO−3结合生成Np(NO3)qn(n=1—6,q=−2—+3)配合物的结构和性质进行了系统研究.
2 理论方法
密度泛函理论利用电子密度代替电子波函数来描述分子体系的物理和化学性质.密度泛函理论的核心是确定一个比较好的交换相关泛函形式,并且将交换泛函和相关联合起来进行计算.本文所使用的是包含梯度修正的B3LYP方法[9−13].对于重元素原子考虑到核电荷数比较大,芯层电子比较多,相对论效果明显,使用常规算法成本较高且时间太长,因此必须采用一个近似方法进行求解.其次,鉴于单原子在形成分子过程中其性质主要由价电子层决定,其内层电子影响并不大.因此,研究中采用相对论原子有效芯势近似.对于元素镎采用斯图加特大学构造的芯层包含60个电子的相对论有效芯势(RECP)[14−16].这种有效芯势用一个“赝势”代替重元素中内层电子与价电子层之间的相互作用,将芯层60个电子以外的n=5壳层(5s,5p,5d,5f),以及6s,6p,6d,7s轨道上的电子当作价电子层处理,所对应的收缩基函数为(12s11p10d8f)/[8s7p6d4f].本组在三卤化镎分子的结构和化学键性质的研究中采用了这种Np的60个电子RECP,通过计算得到NpX3的电荷转移、电子态密度、分子化学键能、光谱和分子稳定性的相关数据,最终计算结果与实验值较为符合.这表明Np采用60个电子的RECP计算所给出的相关数据结果是可信的[17].对于其余元素(N,O)采用6-31+G*全电子基函数[18,19].
计算Np(NO3)qn(n=1—6,q=−2—+3)配合物稳定性的过程实际上是在研究镎离子和硝酸根之间的相互作用强度,需要对镎离子和硝酸根离子的结合能进行计算.在计算过程中使用以下结合能E计算公式:
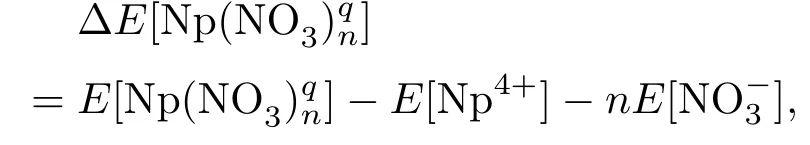
同时,在结合能计算过程中,除了计算方法以及基组对最终的结果有较大影响外,还需考虑基组叠加误差[20],即BSSE.因此对结合能的计算公式修正如下:
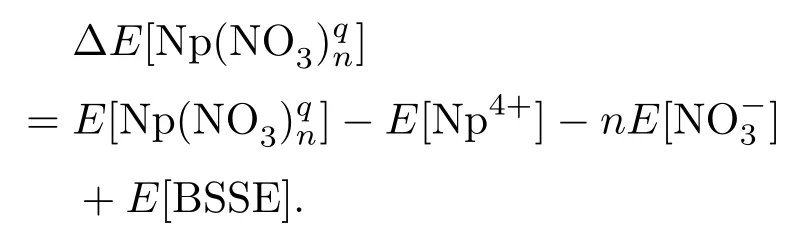
最后,本文的所有计算过程均在Gaussian 03[21]程序中完成,并且所有结构在优化过程中均加入了频率计算,计算结果均无虚频,从而保证了结构体系的稳定性.
3 结果与讨论
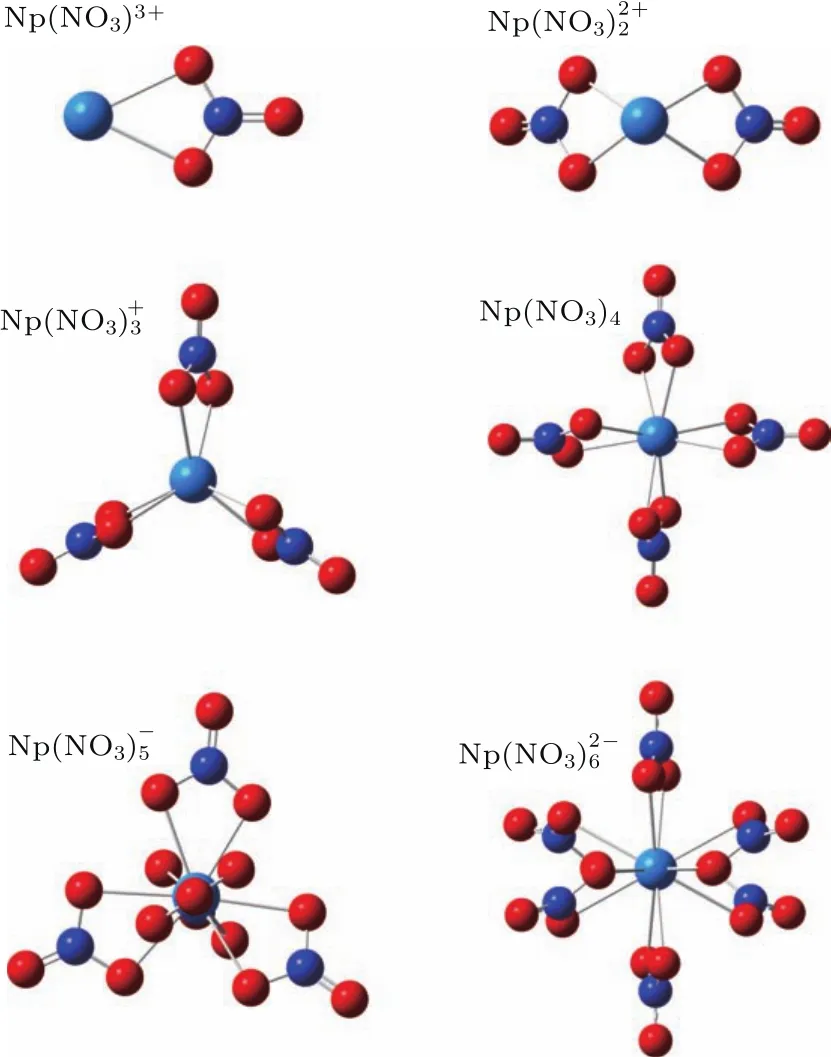
图1 (网刊彩色)配合物的稳定几何结构Fig.1.(color on line)The stab le geom etrical structures of com pounds(n=1–6,q=−2–+3).
硝酸溶液中Np4+与结合可形成(n=1—6,q=−2—+3)配合物.计算中分别优化了Np4+与结合得到的Np(NO3)3+,,, Np(NO3)4,和六种硝酸盐的几何结构,得到的稳定几何结构如图1所示.可以看到,在稳定的几何结构中所有的硝酸根离子与镎离子均以双齿模式结合在一起,且硝酸根离子被镎原子束缚在其周围,并且以对称的形式存在.这表明Np4+与以双齿模式结合时原子之间的相互作用更加强烈,形成的配合物更加稳定.双齿模式的稳定性与施彼得和王金枝[22]在研究Nd与结合形成[Nd(H2O)4(NO)3]2H2O晶体的结论一致(注:以下Oeq均表示上的两个以双齿形式与Np4+结合的氧原子).
在优化的几何结构中,Np(NO3)3+配合物中的Np原子与中的所有原子处在同一平面内.配合物中的Np原子与两个成一个“V”字形,其中 Np原子位于尖端之上,Np原子与N原子间(N—Np—N)组成的夹角为112◦.与Np(NO3)4分子中Np原子与中的N原子均在一个平面上,并且N原子在平面上以一个三角形和矩形的形式存在,Np原子则基本处在三角形和矩形的重心之上,且的位置则都是等价的,在空间以Np原子为原点对称存在.值得注意的是在中,有两个与Np原子在一个轴上,其余四个则是以交叉对称形式存在.
表1 Np(NO3)(n=1—6,q=−2—+3)配合物的稳定结构参数表(单位:键长Å/键角(◦))Tab le 1.The geom etrical param eters of coordination com pound Np(NO3)qn(n=1–6,q=−2–+3)(bond d istance/bond angle()).
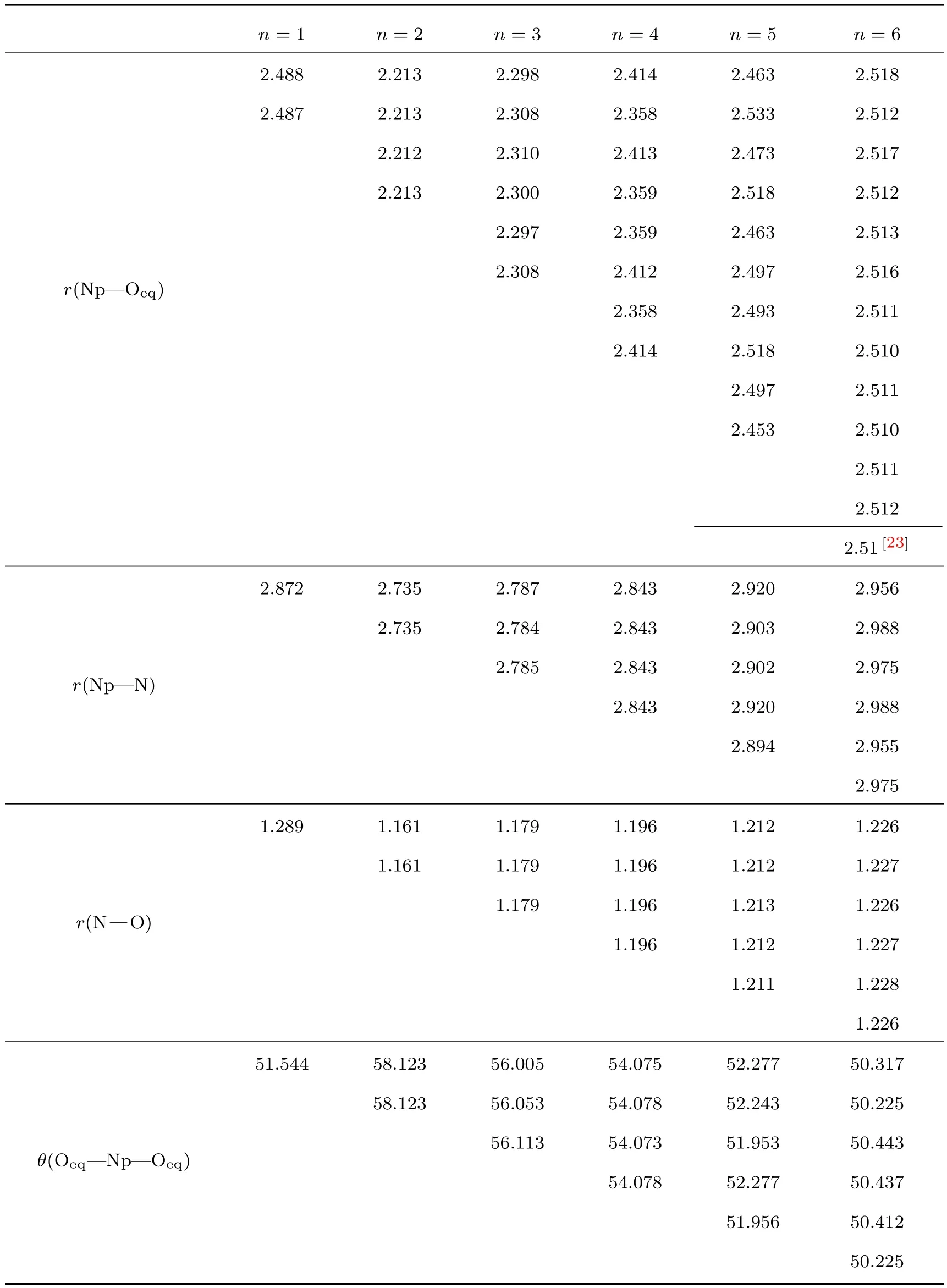
表1 Np(NO3)(n=1—6,q=−2—+3)配合物的稳定结构参数表(单位:键长Å/键角(◦))Tab le 1.The geom etrical param eters of coordination com pound Np(NO3)qn(n=1–6,q=−2–+3)(bond d istance/bond angle()).
n=1 n=2 n=3 n=4 n=5 n=6 r(Np—Oeq) 2.488 2.213 2.298 2.414 2.463 2.518 2.487 2.213 2.308 2.358 2.533 2.512 2.212 2.310 2.413 2.473 2.517 2.213 2.300 2.359 2.518 2.512 2.297 2.359 2.463 2.513 2.308 2.412 2.497 2.516 2.358 2.493 2.511 2.414 2.518 2.510 2.497 2.511 2.453 2.510 2.511 2.512 2.51[23] r(Np—N) 2.872 2.735 2.787 2.843 2.920 2.956 2.735 2.784 2.843 2.903 2.988 2.785 2.843 2.902 2.975 2.843 2.920 2.988 2.894 2.955 2.975 r(N−O) 1.289 1.161 1.179 1.196 1.212 1.226 1.161 1.179 1.196 1.212 1.227 1.179 1.196 1.213 1.226 1.196 1.212 1.227 1.211 1.228 1.226 θ(Oeq—Np—Oeq) 51.544 58.123 56.005 54.075 52.277 50.317 58.123 56.053 54.078 52.243 50.225 56.113 54.073 51.953 50.443 54.078 52.277 50.437 51.956 50.412 50.225
为了研究Np4+在与配体形成配合物过程中是否会影响到配体的结构发生变化,我们通过计算得到配合物(n=1—6,q=−2—+3)中 Np—Oeq与Np—N间的键长以及中两个氧原子与Np原子所组成的键角θ(Oeq—Np—Oeq),与此同时得到配体中N—O键键长,通过结构参数来确定配体的结构变化.表1列出了配合物中Np—Oeq,Np—N间的键长, Oeq—Np—Oeq间的键角θ以及配体中N—O键长的参数大小.例如:Np4+在与结合过程中,配体的N—O间的键长和配合物中的Np—Oeq与Np—N间的键长均发生突变,分别减小0.128Å,0.275Å,0.147Å,之后其键长又平缓的变大.同样,配合物中 θ键角也发生了突变,增大6.579◦,达到最大值为58.12◦,之后随着个数的继续增加θ键角则在逐渐变小.
3.2(n=1—6,q=−2—+3)电荷布居
3.3(n=1—6,q=−2—+3)分子化合物的的结合能
表2 Np(NO3)(n=1—6,q=−2—+3)配合物中的Np原子与硝酸根各原子的电荷转移Tab le 2.Charge transfer of Np atom s in Np(NO3)(n=1–6,q=−2–+3)coordination com pound w ith atom s in nitric acid.
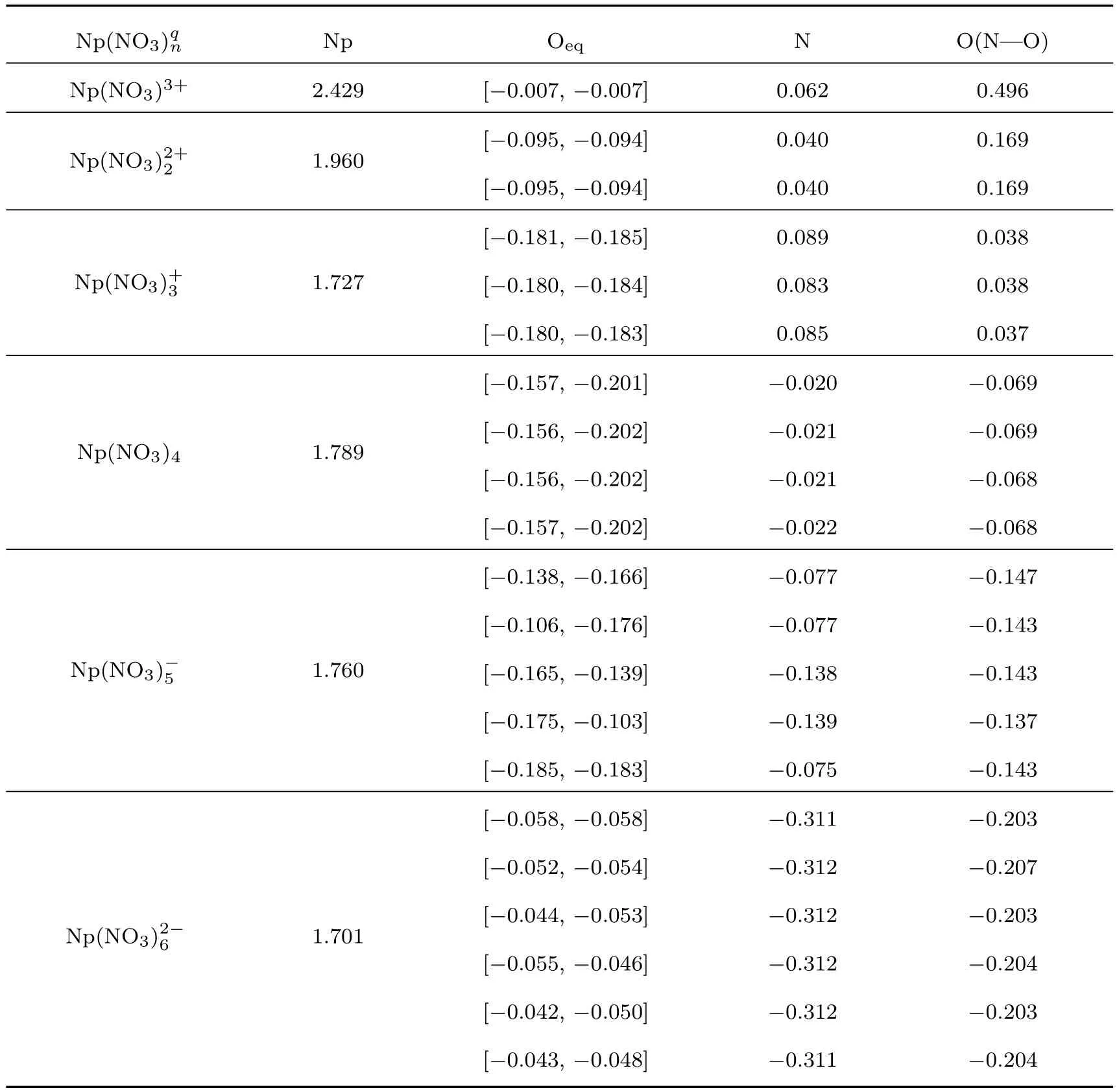
表2 Np(NO3)(n=1—6,q=−2—+3)配合物中的Np原子与硝酸根各原子的电荷转移Tab le 2.Charge transfer of Np atom s in Np(NO3)(n=1–6,q=−2–+3)coordination com pound w ith atom s in nitric acid.
Np(NO3)qn Np Oeq N O(N—O) Np(NO3)3+ 2.429 [−0.007,−0.007] 0.062 0.496 Np(NO3)2+21.960 [−0.095,−0.094] 0.040 0.169 [−0.095,−0.094] 0.040 0.169 Np(NO3)+31.727 [−0.181,−0.185] 0.089 0.038 [−0.180,−0.184] 0.083 0.038 [−0.180,−0.183] 0.085 0.037 Np(NO3)4 1.789 [−0.157,−0.201] −0.020 −0.069 [−0.156,−0.202] −0.021 −0.069 [−0.156,−0.202] −0.021 −0.068 [−0.157,−0.202] −0.022 −0.068 Np(NO3)−51.760 [−0.138,−0.166] −0.077 −0.147 [−0.106,−0.176] −0.077 −0.143 [−0.165,−0.139] −0.138 −0.143 [−0.175,−0.103] −0.139 −0.137 [−0.185,−0.183] −0.075 −0.143 Np(NO3)2−61.701 [−0.058,−0.058] −0.311 −0.203 [−0.052,−0.054] −0.312 −0.207 [−0.044,−0.053] −0.312 −0.203 [−0.055,−0.046] −0.312 −0.204 [−0.042,−0.050] −0.312 −0.203 [−0.043,−0.048] −0.311 −0.204
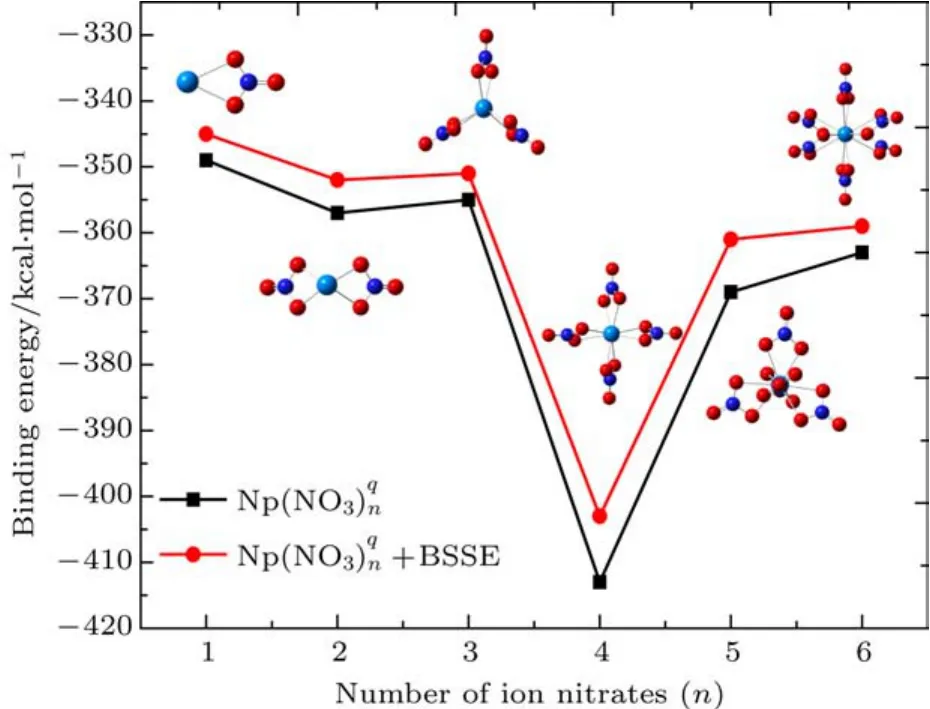
图2 (网刊彩色)Np(NO3)(n=1—6,q=−2—+3)配合物结合能的变化规律Fig.2.(color on line)Bind ing energy of coord ination com pound Np(NO3)(n=1–6,q=−2–+3).
3.4 N p(NO3)4的红外(IR)光谱
IR光谱主要用于化合物鉴定和分子结构的表征,也可以用于定量分析.接下来我们就对稳定结构Np(NO3)4配合物的IR光谱进行讨论.图3为配合物Np(NO3)4在气相和液相条件下的理论计算IR光谱图,在0—1800 cm−1波数范围内,配合物Np(NO3)4有多个IR吸收峰,强的红外振动主要集中在200 cm−1以及1000—1700 cm−1波数范围.光谱中相对强度最大的谱峰出现在1600 cm−1附近,振动强度最大的位置来自于中N—O的伸缩振动.另外,269 cm−1指认为Np—Oeq对称伸缩振动,750,800,1054和1276 cm−1依次分别为N—Oeq剪式振动、摇摆振动、伸缩振动以及反对称伸缩振动.
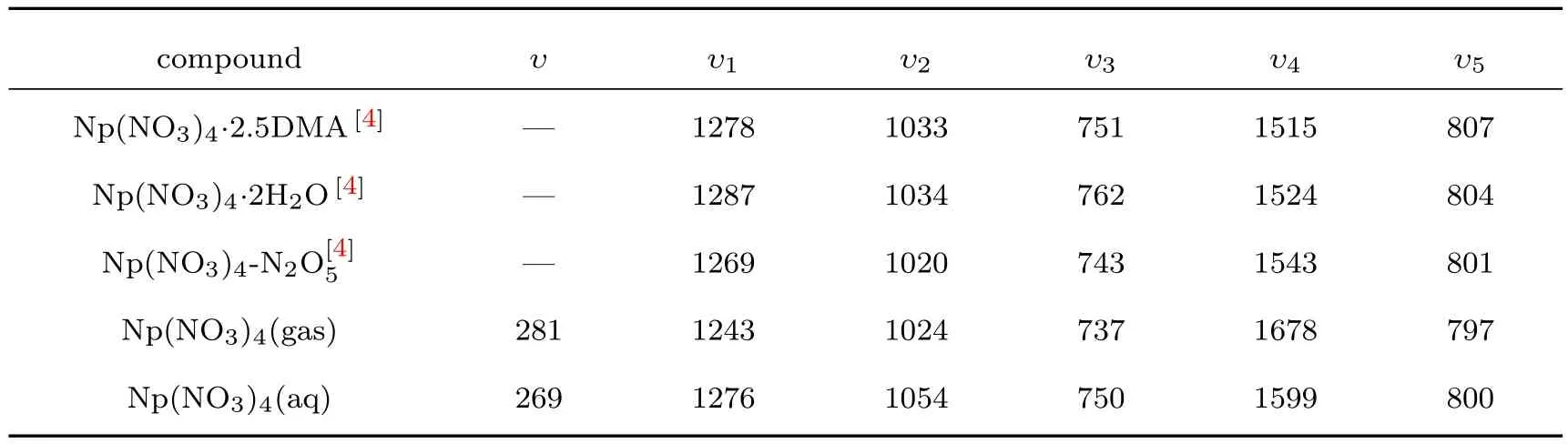
表3 Np(NO3)4配合物在不同溶液中的波数值(cm−1)Table 3.W ave number of coordination com pound in diff erent solutions(cm−1).
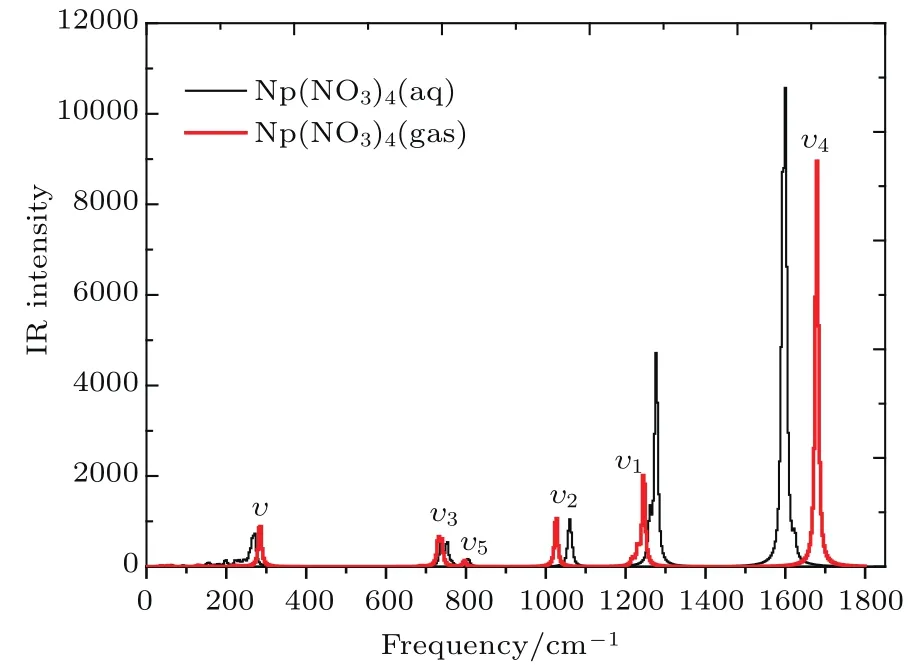
图3 (网刊彩色)计算得到的Np(NO3)4配合物的IR光谱图Fig.3.(color on line)The calcu lated IR spectra for Np(NO3)4com pound.
表3同时列出了Laidler等[4]对Np4+硝酸盐在不同的溶液环境中的IR光谱进行的实验测量结果,实验中测了υ1—υ5的波数.同时,也给出了本文通过理论计算得到的Np(NO3)4在气相和液相条件下相应振动的波数.经过对比不难发现,配合物Np(NO3)4在不同溶液中的υ1,υ2,υ3和υ5波数值的实验结果与理论计算结果符合很好,其中液相条件的计算结果相比气相条件更符合实验结果.但 υ4的计算结果偏较大,这可能是因为υ4作为中N−O的伸缩振动,其O(N−O)原子离Np原子最远,受溶液环境的影响较大.
4 结 论
本文基于密度泛函理论的Gaussian 03程序,系统研究了IV价镎离子(Np4+)与不同个数的硝酸根离子结合形成的配合物(n=1—6, q= −2—+3)的结构和性质. 研究结果表明,配合物以双齿形结合为稳定的结合形式;其次,在一般情况下,键长短、键能大,破坏该键就困难,物质就越稳定.在本文中Np4+与之间形成的配合物中Np与中的N,Oeq原子间的距离最短.但是根据结合能的计算,Np4+更倾向于结合形成稳定的配合物Np(NO3)4.最后对稳定结构Np(NO3)4配合物进行IR光谱计算,并且与实验数据进行对比,结果符合比较好.
[1]Zhou W G 2011 Prog.In.Chem.23 1272(in Chinese) [韦悦周2011化学进展23 1272]
[2]T ian G X 2015 J.Nucl.Radioana l 37 276(in Chinese) [田国新2015核化学与放射化学37 276]
[3]W u Y P 1996 Atom.En.Sci.Technol.30 179(in Chinese)[吴宇平1996原子能科学技术30 179]
[4]Laid ler J B 1966 J.Chem.Soc.A 88 780
[5]Sergei M ikhailov B A(translated by Zhang X X,Zhu J,Zhao S Z)1971 Neptunium in Analytical Chem istry (Beijing:Atom ic Energy Press)pp19–23(in Chinese)[米哈伊诺夫B A著(张心祥,祝疆,姜延林,赵守真 译)1971镎的分析化学(北京:原子能出版社)第19—23页]
[6]Y in Y P,Dong C Z,D ing X B 2015 Chem.Phys.Lett. 635 134
[7]Zeng J H,Yang X,Liao J L,Liu N,Yang Y Y,Chai Z F,W ang D Q 2014 Phys.Chem.Chem.Phys.16 16536
[8]Yang X,Liang Y N,D ing S D,Li S J,Chai Z F,W ang D Q 2014 Inorg.Chem.53 7848
[9]Vallet V,M acak P,W ah lgren U,G renthe I 2006 Theor. Chem.Acc.115 145
[10]Lee C,Yang W,Parr R G 1988 Phys.Rev.B 37 785
[11]Schreckenbach G,Hay P J,M artin R L 1999 J.Com put. Chem.20 70
[12]Eh lers A W,Frenking G 1994 J.Am.Chem.Soc.116 1514
[13]Zheng Y Y,Ren G M,Chen R,W ang X M,Chen X H, W ang L,Yuan L,Huang X F 2014 Acta Phys.Sin.63 213101(in Chinese)[郑圆圆,任桂明,陈锐,王兴明,谌晓洪,王玲,袁丽,黄晓凤2014物理学报63 213101]
[14]Cao X,Dolg M,Stoll H 2003 J.Chem.Phys.118 487
[15]Cao X,Dolg M,Stoll H 2004 J.M olec.Struct. (Theochem)673 203
[16]K uch le W,Dolg M,Stoll H,Preuss H 1994 J.Chem. Phys.100 7535
[17]Y in Y P,Dong C Z,Du L Q 2014 Eur.Phys.J.D 68 1
[18]Hay P J,M artin R L 1998 J.Chem.Phys.109 3875
[19]G lukhov tsev M N,Pross A,M cgrath M P 1995 J.Chem. Phys.103 1878
[20]Boys S F,Bernardi F 1970 M ol.Phys.19 553
[21]Frisch M J,Trucks G W,Sch legel H B,Scuseria G E, Robb M A,Cheesem an JR,M ontgom ery Jr J A,Vreven T,K ud in K N,Burant J C 2004 Gaussian 03 Revision B.05(W allingford:Gaussian Inc)
[22]Shi B D,W ang J Z 1991 J.X iam en Univ.1 55(in Chinese)[施彼得,王金枝1991厦门大学学报(自然科学版)1 55]
[23]A llen P G,Bucher J J,Shuh D K,Edelstein N M,Reich T 1997 Inorg.Chem.36 4676
PACS:31.10.+z,31.15.E–,31.15.esDOI:10.7498/aps.66.063101
Structu res and p roperties of N p(NO3)(n=1–6, q=−2–+3)coord ination com pound∗
Ma Lei Yin Yao-Peng Ding Xiao-Bin Dong Chen-Zhong†
(Key Laboratory of A tom ic and M olecu lar Physics and Functional M aterials of Gansu Province,College of Physics and E lectronic Engineering,Northw est Norm al University,Lanzhou 730070,China)
(Received 13 October 2016;revised m anuscript received 8 Decem ber 2016)
In the process of nuclear waste disposal,the valuab le uranium and p lutonium are recycled and separated by dissolving the spent fuel in nitric acid.However,transuranic Np greatly influences the process of separation and recovery. Therefore,it is vital to study the structure and properties of nitrate,which is combined w ith neptunium ions and nitric acid.Furtherm ore,there are few researches about nitrate form ed by tetravalent neptunium ions.So in this article,by using B 3LYP hybrid m ethod of density functional theory,the Gaussian 03 program is used to optim ize the geom etric construction of the coordination com pounds(n=1–6,q=−2–+3)formed by the tetravalent neptunium ions(Np4+)and nitrate ion.Under the relativistic eff ective core potentialmodel,the structure param eters and properties are reported.It is found thatcoordinates to Np4+as a bidentate ligand,and the Np—N and Np—O bonds are the shortest in,while the binding energy of the Np(NO3)4is the largest.The infrared spectra of Np(NO3)4are calculated in the gas and liquid phase.Com paring w ith the available experim ental data,the reliability of the calcu lation results in thiswork is confi rmed.
,density functional theory,binding energy,infrared spectral analysis
10.7498/aps.66.063101
∗国家自然科学基金重大研究计划(批准号:91126007)资助的课题.
†通信作者.E-m ail:dongcz@nw nu.edu.cn
*Pro ject supported by the M a jor Research P lan of the National Natural Science Foundation of China(G rant No.91126007).
†Corresponding author.E-m ail:dongcz@nwnu.edu.cn
猜你喜欢
大学物理(2022年9期)2022-09-28
工业催化(2020年9期)2020-11-13
无机化学学报(2020年7期)2020-07-20
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
无机化学学报(2018年8期)2018-08-01
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
原子与分子物理学报(2015年3期)2015-11-24
郑州大学学报(理学版)(2012年4期)2012-03-25
