
二代测序在遗传性神经肌肉病分子生物学中的应用
2017-07-19 13:48卢正娟常蕾蕾吴家勇牛丰南综述运审校
中风与神经疾病杂志 2017年6期
赵 燕, 卢正娟, 刘 卓, 常蕾蕾, 吴家勇, 牛丰南综述, 徐 运审校
二代测序在遗传性神经肌肉病分子生物学中的应用
赵 燕1, 卢正娟1, 刘 卓1, 常蕾蕾1, 吴家勇1, 牛丰南2综述, 徐 运1审校
遗传性神经肌肉病(inherited neuromuscular disorders,NMDs)是一组少见的累及神经(遗传性运动感觉周围神经病、肌萎缩侧索硬化、遗传性脊肌萎缩症等)、肌肉(各型肌病/肌营养不良、离子通道病等)及神经-肌肉接头疾病的单基因遗传性疾病,多以肌无力、肌萎缩,伴/不伴肌张力低下及感觉障碍为主要临床表现,可于新生儿、儿童或成年期发病,为患者及其家庭以及公共卫生造成极大负担。该类疾病具有临床/遗传异质性,基因检测是诊断的金标准。以往Sanger法一直是DNA测序的最主要方法和金标准。然而不典型的临床症状、庞大的致病基因谱及致病基因分子量巨大,导致传统测序方法对该类疾病诊断具有局限性。2005年,Roche公司的454测序仪,可以进行大规模平行焦磷酸测序,标志着高通量基因组分析时代的到来。接着Illumina公司的Solexa技术以及ABI公司的SOLiD技术为标志各种新测序平台和技术的逐渐成熟,宣告新一代测序技术时代的来临[1]。大规模平行测序技术(massively parallel sequencing,MPS),又称下一代测序技术(next generation sequencing,NGS),可以快速诊断大部分NMDs。
1 NGS的概述
1.1 NGS与一代测序比较 DNA测序技术诞生于上世纪70年代,逐渐成为单基因遗传病确诊的最直接和准确的手段。几十年来,经典的DNA测序技术的发展经历了Maxam-Gilbert化学测定、Sanger链终止测序等阶段,以毛细管电泳为代表的自动化Sanger测序又称为第一代测序技术,并成为此后20年间被广泛应用的DNA测序技术。目前Sanger测序可以一次检测最大长度约1 kb的DNA片段,准确度达到99.999%。虽然经典Sanger测序对人类遗传性疾病的诊断做出了很大贡献,但该方法测序效率低而成本高,对巨大基因、染色体甚至整个基因组的测序费时费力。此外Sanger法测序是先合成再测序,测序前必须先扩增目的片段。如果进行DNA重组生物体内扩增,则成本高,耗时长;PCR体外扩增,由于PCR时各种条件限制,引物设计等多方面因素,会导致无法扩增出目的片段,即使能够扩增目的片段,可重复性差,无法稳定扩增目的片段。
NGS技术特点是边合成边测序(通过捕捉新合成的末端的标记来确定DNA的序列),而且通量高、速度快、单碱基成本低,与传统的Sanger测序相比,NGS在各个遗传学领域均具有革命性的进步。其实验主要分为以下几个步骤[2]:(1)基因组DNA随机片段化、测序文库构建及文库片段加测序接头;(2)外显子杂交捕获、富集(全基因组测序时所有的DNA片段不需捕获均行NGS检测);(3)外显子扩增和NGS测序;(4)生物信息学分析。由于采用并行测序,可同时对数以万计的DNA片段进行测序,测序效率较经典Sanger测序提高了成千上万倍,同时单位测序量的成本仅为Sanger测序的万分之一;通过反复测序同一区域的DNA片段达到Sanger测序所不能企及的灵敏度及准确度[2]。
NGS技术与传统的Sanger测序相比较,尽管它的高通量减少了成本,但是读取准确性以及片段的长度都不及Sanger测序,这些不足都可以通过测序的覆盖率和生物信息学的方法来克服。
1.2 NGS的几大平台及应用 目前主流的NGS平台有Illumina公司的HiSeq2000系列、罗氏454系列以及LifeTech公司的SOLiD系列、Polonator平台。这几个技术平台各有优点,454测序片段长,Solexa测序性价比最高,SOLID测序准确度高。上述测序平台成本较高,通量太大,实验周期较长,主要应用于基础研究领域或重大项目的大规模测序之中,并不适合于医疗领域的分子检测、临床诊断方面。为了满足医院及中小实验室的测序需求,三大公司相继都推出了低通量、低成本的测序平台:Life Tech的IonTorrent(PGM、Proton)、Illumina的Miseq以及Roche的GS Junior(见表1)。
1.3 NGS生物信息分析流程 主要包括:(1)质量控制:对测序产生的原始数据(Raw data)进行质控、过滤低质量序列等处理,得到Clean data;(2)将Clean data比对到参考基因组;(3)将比对好的文件进行突变检测,使用不同的软件检测SNVs和InDel,WGS还可使用多种软件检测CNVs;(4)注释突变及筛选致病基因:利用ANNOVAR对检测到的成千上万的突变进行注释(突变对蛋白质编码的情况及突变位于各基因区间信息等),利用已知突变数据库如dbSNP、1000 Genome、ESP 6500等,去除出现频率较高的突变,利用预测软件(SIFT、Polyphen等)对突变进行致病性和保守性预测,同时通过疾病数据库将突变与疾病表型联系起来。
1.4 NGS在医学上的主要应用 NGS的应用十分广泛,包括基因组测序、转录组测序和表观遗传测序、DNA和蛋白质相互作用研究以及基因组DNA甲基化分析等,而在孟德尔遗传型疾病中的应用主要为基因组测序,包括如下两大类: (1)全基因组重测序(Whole-genome sequencing,WGS):是指对某物种的整个基因组进行高通量测序。通过WGS能够全面检测个体基因组上所有的基因突变[3],其在测序过程中不需要捕获外显子,对基因组所有DNA片段进行测序。WGS可检测人全基因组水平与疾病相关的单核苷酸变异(SNVs)、插入缺失(InDels)、拷贝数变异(CNV)和结构变异(SV)及非编码区的SNVs、InDels等多种全面的突变信息[4,5]。WGS的缺点是价格昂贵、数据量巨大而使数据分析速度慢及测序深度较低和覆盖度不足而使测序精度不能达到临床应用的标准[6]。(2)目标区域测序:利用特制探针对感兴趣的基因组区域进行富集,结合NGS技术将这些富集的目标区域进行测序。目标测序法由于其测序的目标区域只占全基因组的小部分,相比WGS能够达到很高的测序深度。主要分为以下两种:(1)外显子测序(Whole-exome sequencing,WES):将基因组外显子区域DNA利用序列捕获技术进行捕获,然后进行高通量测序。外显子区域仅占人类基因组区域的1%左右,却拥有约85%与疾病相关的变异位点[7]。因此,对外显子区域选择性的测序,即能检测大部分人类疾病致病基因及突变。有研究认为,如果不考虑价格因素,WGS可以比WES更有效地检测外显子组的突变。(2)目标区域测序(targeted regions sequencing,TRS)指将感兴趣的基因区域DNA利用序列捕获技术进行捕获,然后进行高通量测序。与外显子测序相比,目的基因富集测序,对已知疾病基因谱系全面筛查,相比外显子测序,更为高效,覆盖度更全面,测序深度更有保障,以及测序费用最为低廉,因此用于临床最为广泛[8~10]。 捕获测序在研究单基因遗传病和多基因遗传疾病(如癌症、糖尿病、肥胖症等)的致病基因和易感基因方面起到极为重要的作用,为疾病的临床分子诊断提供了新的线索。
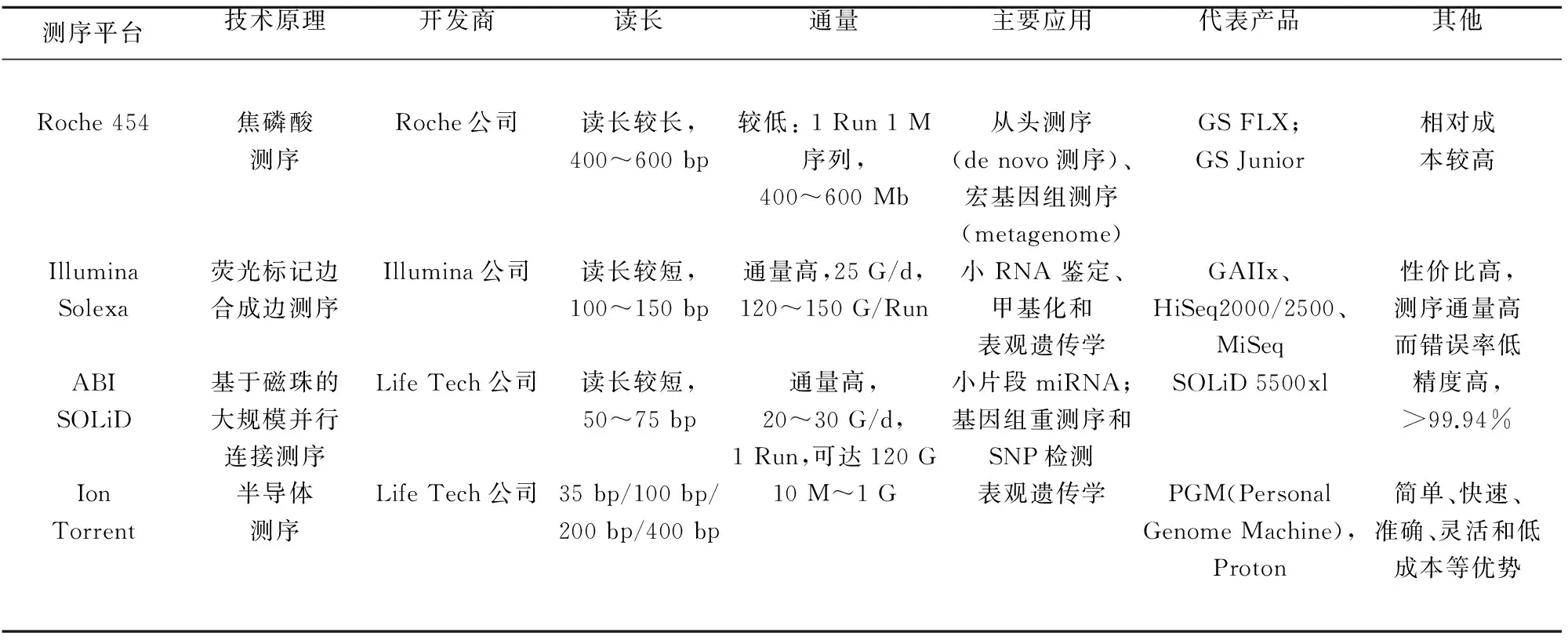
表1 二代测序主流的四大技术平台
2 高通量测序技术在NMDs的应用
2.1 该组疾病的分子诊断存在诸多挑战
2.1.1 遗传异质性 NDMs是一组最具遗传异质性的疾病,涉及基因数目达300多种。如CMT有30多种致病基因,肢带型肌营养不良致病基因20多种等。
2.1.2 大基因的涉入 NDMs包含了众多人类最大的致病基因,如DMD致病基因DMD(MIM#300377),有79个外显子,分子量超过2.3 Mb[11];先天性肌病基因谱系中TTN(MIM#188840)拥有363个外显子[12];NEB(MIM#161650)有183个外显子;而RYR1(MIM#180901)有106个外显子。如果检测这些基因,以往的Sanger测序只是筛查热点突变或部分区域,很少对整个基因进行测序。
2.1.3 临床异质性 同种基因变异可以引起多种疾病表型。如RYR1为多种先天性肌病(中央轴空病、多微小轴空病、肌纤维类型不均、中心核肌病)及其他肌病(恶性高热、King-Denborough综合征)的致病基因[13~17]。而且同一种突变亦可导致严重程度不等的临床表现,因此难以通过临床表现确定可能的致病基因。
2.1.4 临床特点不明确 很大一部分患者临床表现及骨骼肌病理无明显特征,如先天性肌病中根据骨骼肌病理活检通常分为杆状体肌病、轴空性肌病及中心核肌病等,然而近一半的患者并没有这些特殊表现,而有特征表现的患者,还有临床及病理现象重叠的表现。因此传统的一代测序法测出某一候选基因的突变即会终止实验,而导致未能筛选出真正的致病基因,若候选基因未检测到突变,则需要费时费力的挨个的筛查其他基因。
2.1.5 需要使用不同的技术 NMDs患者的突变具有多种形式,例如DMD约60%~65%存在缺失;5%~15%为重复突变;剩余的为点突变及小的插入缺失突变[18],因此分子诊断需要不同的实验技术来实现,如MLPA。
2.1.6 未明确的基因 很多NMDs研究不明确,存在未知的致病基因,如肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)可能与以下基因有关:SOD1 (MIM#147450)、SETX (MIM#608465)、TARDBP (MIM#605078)[19]、FUS (MIM#137070)等[20],然而大部分ALS患者在以上基因中均未发现突变,提示可能存在其他致病基因。综合以上因素约40%的NMDs患者不能获得最终的分子诊断。
2010年,Lupski[21]等第一次报道了在NMDs运用NGS技术,他们运用CMT相关基因的全外显子组确定1例AR遗传的CMT为SH3TC2的复合杂合突变。此后,NMDs运用大规模平行测序技术检测可能的致病突变的报道越来越多。
2012年,Vasli等[22]利用靶向基因富集(267个NMDs相关基因)外显子组测序对16例NMDs患者进行检测,8例有明确临床表型,找到了致病,同时也在另8例临床表现相似和遗传缺陷未知的NMDs患者中的5例样本中找到了候选致病突变。Vasli进一步认为,在对此类患者采取侵入性或其他特殊手段进行诊断性试验之前,将NGS技术作为常规诊断试验进行候选基因突变筛查,对于遗传性神经肌肉疾病患者的临床诊疗和遗传咨询具有重要价值。Lim等则指出NGS实验中某些编码区域测序深度不够可能使其检测灵敏度降低,仍需采用Sanger测序进行补充。
随着生物信息学的飞速发展,使用NGS进行CNVs的诊断已经得到实践[23]。越来越多的报道证实NGS在诊断DMD/BMD中的广泛应用,其既可以检测CNVs,又可同时筛查点突变,大大提高检测效率[24]。
NGS具有其技术局限性,在核苷酸序列重复疾病如强直性肌营养不良、脊髓小脑共济失调、肯尼迪病等,不如传统片段分析法准确、低廉,但是随着捕获技术及生物信息学技术的进步,NGS的应用会越来越广泛[25]。
3 NGS的局限性及展望
3.1 假阴性 某些NMDs可能有重复片段导致假阴性结果,可以增加测序覆盖度、提高生物信息分析技术及更新生物信息分析软件,必要时结合其他分子检测技术(如毛细管电泳片段分析)共同诊断。在目标区域捕获测序中,可以提高捕获探针数目,以增加基因编码区的覆盖度降低假阴性率。
3.2 假阳性 通过NGS途径可产生成百上千SNV及InDels,数据分析没有统一标准,另外测序时重复区域的碱基延伸具有一定错误率,造成假阳性结果,后者的错误可通过Sanger测序进行验证,但前者错误仍是NGS应用的最大限制。
3.3 海量数据的存储及处理 如标准的WGS测序数据可达100 GB左右,若经过生物信息分析后可得到clean data、BAM文件、SAM 文件,存储空间需要300 GB。而如此巨大的数据在数据的管理、分析处理及存储均带来极大挑战。目前快速发展的云存储、云计算为这一问题带来了曙光。
综上NGS技术平台的可以一次对多个位点或者同样的位点进行深度检测,检测到基因结构的低频变化,在NMDs诊断中提供较为全面和深度的信息,可代替大部分基因测序方法,但不能替代临床检查及骨骼肌活检。临床及骨骼肌病理可指导基因测序方向。少部分NMDs,具有明确表型和有家族史的患者,拒绝/无条件有创检查(骨骼肌活检等)时,可以直接行NGS检测[26]。总之,NGS在NMDs的临床诊断及研究中发挥重要作用。
[1]Reinke T.Next-generation sequencing problematic,if promising[J].Managed Care,2013,22(12):38-41.
[2]Mueller W,Lyons J,Kerr G,et al.Standard enrichment methods for targeted next-generation sequencing in high-repeat genomic regions[J].Genetics Med,2013,15(11):910-911.
[3]Schnekenberg RP,Nemeth AH.Next-generation sequencing in childhood disorders[J].Archiv Dis Childhood,2014,99(3):284-290.
[4]Brunstein J.High throughput sequencing:next generation methods[J].MLO:Medical Laboratory Observer,2013,45(10):36,8-9.
[5]Parson W,Strobl C,Huber G,et al.Reprint of:Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM)[J].Forensic Sci Inter Genetic,2013,7(6):632-639.
[6]Korfhage C,Fisch E,Fricke E,et al.Whole-genome amplification of single-cell genomes for next-generation sequencing[J].Current Protocols Molec Biol,2013,104(Unit):7-14.
[7]Ross LF,Rothstein MA,Clayton EW.Premature guidance about whole-genome sequencing [J].Personalized Med,2013,10(6):523-526
[8]Menzies D.Molecular methods for tuberculosis trials:time for whole-genome sequencing[J].Lancet Respiratory Med,2013,1(10):759-761.
[9]Mamanova L,Coffey AJ,Scott CE,et al.Target-enrichment strategies for next-generation sequencing [J].Nature Methods,2010,7(2):111-118.
[10]Choi M,Scholl U I,JW,et al.Genetic diagnosis by whole exome capture and massively parallel DNA sequencing[J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(45):19096-190101.
[11]Garcia-Planells J,Torres-Puente M,Vilchez JJ,et al.Novel human pathological mutations.Gene symbol:DMD.Disease:muscular dystrophy,Duchenne[J].Hum Genet,2009,126(2):338.
[12]D’Argenio V,Frisso G,Precone V,et al.DNA sequence capture and next-generation sequencing for the molecular diagnosis of genetic cardiomyopathies[J].J Molecular Diagnostics,2014,16(1):32-44.
[13]Bertini E,Darras BT.Congenital myopathies:rebuilding the natural history,one gene at a time[J].Neurology,2015,84(1):15-16.
[14]Lillis S,Abbs S,Mueller CR,et al.Clinical utility gene card for:Central core disease[J].European J Human Genetics,2012,20:2.
[15]Amburgey K,Bailey A,Hwang JH,et al.Genotype-phenotype correlations in recessive RYR1-related myopathies[J].Orphanet J Rare Dis,2013,8:117.
[16]Dowling JJ,Lillis S,Amburgey K,et al.King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene[J].Neuromuscular Disord,2011,21(6):420-427.
[17]Bevilacqua JA,Monnier N,Bitoun M,et al.Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization[J].Neuropathol Applied Neurobiol,2011,37(3):271-284.
[18]White SJ,den Dunnen JT.Copy number variation in the genome; the human DMD gene as an example[J].Cytogenetic Genome Res,2006,115(3/4):240-246.
[19]Sheerin UM,Schneider SA,Carr L,et al.ALS2 mutations:juvenile amyotrophic lateral sclerosis and generalized dystonia[J].Neurology,2014,82(12):1065-1067.
[20]Birve A,Neuwirth C,Weber M,et al.A novel SOD1 splice site mutation associated with familial ALS revealed by SOD activity analysis[J].Human Molecul Genetic,2010,19(21):4201-4426.
[21]Lupski JR,Reid JG,Gonzaga-Jauregui C,et al.Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy[J].New Eng J Med,2010,362(13):1181-1191.
[22]Vasli N,Bohm J,Le Gras S,et al.Next generation sequencing for molecular diagnosis of neuromuscular diseases[J].Acta Neuropathol,2012,124(2):273-283.
[23]Lee KW,Woon PS,Teo YY,et al.Genome wide association studies (GWAS) and copy number variation (CNV) studies of the major psychoses:what have we learnt[J].Neurosci Biobehavioral Rev,2012,36(1):556-571.
[24]Niba ET,Tran VK,Tuan-Phamle A,et al.Validation of ambiguous MLPA results by targeted next-generation sequencing discloses a nonsense mutation in the DMD gene[J].Clinica Chimica Acta,2014,436:155-159.
[25]Ellard S,Patrinos GP,Oetting WS.Clinical applications of next-generation sequencing:the 2013 human genome variation society scientific meeting[J].Human Mutation,2013,34(11):1583-1587.
[26]Johansen Taber KA,Dickinson BD,Wilson M.The promise and challenges of next-generation genome sequencing for clinical care[J].JAMA Internal Med,2014,174(2):275-280.
1003-2754(2017)06-0569-03
R746
2016-12-11;
2017-01-29
国家自然科学基金青年基金(No.81300977);国家自然科学基金面上项目(No.81671113)
(1.南京大学医学院附属鼓楼医院神经内科,江苏 南京 210008;2.南京大学医学院附属鼓楼医院病理科,江苏 南京 210008)
徐 运,E-mail:xuyun20042001@aliyun.com
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
军事文摘(2022年16期)2022-08-24
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
湖北农业科学(2014年11期)2014-09-10
