
顶空气相色谱法快速测定酒中氰化物含量
2017-07-18 11:57杜利君刘晓林高媛惠郭丽华刘海峰李菲菲
食品研究与开发 2017年11期
杜利君,刘晓林,高媛惠,郭丽华,刘海峰,李菲菲
(山西出入境检验检疫局技术中心,山西太原030024)
顶空气相色谱法快速测定酒中氰化物含量
杜利君,刘晓林,高媛惠,郭丽华,刘海峰,李菲菲
(山西出入境检验检疫局技术中心,山西太原030024)
该文建立了顶空气相色谱法测定酒中氰化物含量的方法。酒样品用氯胺T将氰化物衍生为氯化氰,顶空进样,经WAX气相色谱柱(30 m×0.32 mm×0.50 μm)分离,电子捕获检测器(ECD)检测,外标法定量。同时对检测过程中的衍生剂用量、衍生酸度、顶空平衡温度、顶空平衡时间、气相色谱条件进行优化,结果表明:氰离子在0.001 mg/L~0.1 mg/L 范围内线性良好,相关系数可达到 0.998,定量限(以信噪比(S/N)≥10)为 0.05 mg/L。在 0.1、0.2、8.0 mg/L 3个添加水平下进行了回收率和精密度试验,其中加标回收率在84.1%~109.7%之间,相对标准偏差RSD(n=10)为4.41%~6.54%,满足日常的检测要求。与传统的分光光度法相比,该方法操作简便、快速、稳定性好、灵敏度高、抗干扰能力强,在实际工作中有很好的应用价值。
酒;顶空气相色谱法;氰化物
氰化物是一类带有氰基(CN-)的剧毒化合物,主要指氰酸盐和氢氰酸[1]。可通过呼吸道、消化道及皮肤进入人体内。其毒性主要由氰化物在体内解离出的CN-引起,CN-与细胞色素氧化酶aa3中的Fe3+结合,使酶丧失活性,导致细胞内呼吸中断,阻断电子传递和氧化磷酸化,从根本上抑制三磷腺苷的合成,从而抑制了细胞内氧的利用,机体缺氧从而产生中毒效应[2]。
食品安全是国家和公众普遍关注的问题,而氰化物是酒产品的一项重要安全检测指标。我国作为酒类产品消费大国,建立一种快速、灵敏的检测酒中氰化物的方法具有重大意义。GB 2757-2012《食品安全国家标准蒸馏酒及其配制酒》[3]中规定,酒中氰化物含量不得超过8 mg/L(以HCN计)。目前,检测氰化物的方法主要有分光光度法[4-9]、电化学法[10]、气相色谱法[11-12]、离子色谱法[13]、气相色谱质谱法[14]。其中国标法GB/T 5009.48-2003《蒸馏酒与配制酒卫生标准的分析方法》[15]推荐异烟酸-吡唑啉酮分光光度法测定酒中氰化物,氰化物在酸性溶液中蒸出后,被吸收于碱性溶液中,在中性介质中氰化物与氯胺T反应生成氯化氰(CNCl),再与异烟酸反应,经水解生成戊稀二醛,戊稀二醛与吡唑啉酮缩合产生蓝色染料。该方法对反应体系pH要求较严,而对用来调节pH的酸、碱使用量又限制得非常小(不到0.8 mL)。在实际操作时,常常会超出所限体积,从而导致试验失败,而对于浑浊或有色的酒需要先进行蒸馏,增加了检测工作量,即使经过蒸馏在显色的过程中也易出现浑浊,造成比色困难,影响检测结果的准确性。
本文将氰化物用氯胺T衍生为氯化氰后,直接顶空进样,气相色谱法ECD检测器检测,样品不需蒸馏,操作简便快速,干扰少,实用性强,应用该方法对市售的白酒进行检测,获得了满意的结果。
1 试验部分
1.1 仪器与试剂
GC2010气相色谱仪(配有ECD检测器):日本岛津公司;VortexGenie2旋涡混合器:美国Vor-tex-Genie公司;TurboMatrix HS自动顶空进样器:美国PerkinElmer;超声波清洗器:昆山市超声仪器有限公司。
水中氰成分分析标准物质(50 μg/mL):中国标准物质网;磷酸、氢氧化钠、氯胺试剂T(分析纯):天津市科密欧化学试剂有限公司;试验用水为超纯水:美国密理博公司。
1.2 试验方法
1.2.1 溶液配制
氰离子标准工作溶液:使用时将水中氰成分分析标准物质用水稀释成质量浓度为 0、0.001、0.002、0.010、0.050、0.100 mg/L的标准工作溶液。准确移取10 mL标准工作溶液于顶空瓶中,涡流混合,然后加入0.2 mL10 g/L的氯胺T溶液,立即加盖密封,涡流混合,待测。
1.2.2 样品处理
准确移取0.2 mL试样于顶空瓶中,加入蒸馏水9.8 mL,涡流混合,然后加入0.2 mL 10 g/L的氯胺T溶液,立即加盖密封,涡流混合,待测。
1.2.3 气相色谱条件
色谱柱:WAX毛细管柱(30 m×0.32 mm×0.50 μm);色谱柱温度:40℃保持5 min,以50℃/min速率升至200℃保持2 min;载气:氮气;纯度不低于99.999%;进样口温度:200℃;检测器温度:260℃;分流比为5∶1;柱流速:2.0 mL/min。
1.2.4 顶空条件
顶空平衡温度:50℃;取样针温度:55℃;传输线温度:100℃;顶空平衡时间:30 min;进样时间:0.03 min;加压时间:1 min;载气压力:0.176 MPa。
2 结果与讨论
2.1 样品前处理条件的选择
由于本方法采用气相色谱法检测,溶液的浑浊和颜色都不会对检测造成影响,所以直接取酒样进行检测。
2.2 衍生剂用量的选择
氰离子不能被气相色谱直接检测,需经过衍生处理转化为具有挥发性和稳定性的衍生物,从而实现气相色谱分析。其中氯胺T衍生法为反应生成氯化氰,氯化氰具有沸点低、易挥发的特点,可以实现顶空进样检测。顶空技术的应用大大降低了对样品的前处理过程,减少了样品本身可能对分析的干扰或污染。同时顶空分析技术对分析人员和环境危害小、方法重现性好、能实现自动化。
氯胺T作为衍生剂,它的用量对检测结果影响较大,将顶空瓶中氰离子的浓度固定在0.1 mg/L,分别添加 0.01、0.02、0.05、0.1、0.2、0.3、0.5 mL 的 10 g/L 的氯胺T溶液,观察氯胺T的用量对色谱响应值的影响。由试验数据可以看出氯胺T用量达到50 μL后峰面积稳定,即可获得满意的结果。但氯胺T的在存放过程中容易分解而降低活性氯的含量,所以为保证有足够的氧化能力,选取衍生剂用量为0.2 mL。
2.3 衍生酸度的选择
根据文献得知,衍生是在中性和酸性条件下进行的,因此通过在顶空瓶中加入浓磷酸 0、10、20、30、40、50、100、200 μL 达到调节 pH 值的目的,观察对色谱响应值的影响,发现溶液pH值在2~6范围内时没有明显差别。所以本方法直接用超纯水稀释酒样进行检测。
2.4 顶空平衡温度选择
选择顶空平衡温度为 35、45、50、55、65、75 ℃共6个温度进行试验,发现在35℃~50℃时,响应值随着温度的升高而升高,当温度达到50℃后变化不大,由于温度过高会导致进样的杂质增大,因此最终选择50℃为顶空平衡温度。
2.5 顶空平衡时间的选择
对0.2 mg/L的标准溶液的平衡时间选择10、20、30、40、50 min分别进行考察,发现顶空平衡时间30 min后即达到稳定。故最终平衡时间选择为30 min。
2.6 线性范围的考察
分别配制氰离子浓度为 0、0.001、0.002、0.005、0.01、0.05、0.1 mg/L系列标准溶液进行气相色谱测定,以峰面积(Y轴)对氰化物的衍生物的浓度(X轴)绘制标准曲线。氰化物在0.001 mg/L~0.1 mg/L范围内有良好的线性关系,线性方程为:y=3×107x+52 596,相关性系数为0.998。本方法通过添加回收试验确定检出限0.02 mg/L,定量限0.05 mg/L。在上述条件下,氰离子衍生物的色谱图见图1。
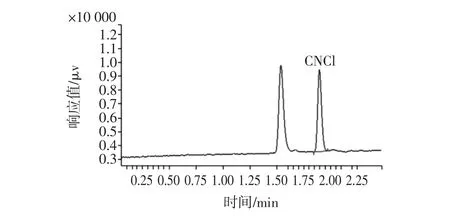
图1 氯化物衍生物标准色谱图(0.01 mg/L)Fig.1 Chromatogram of the thiocyanate standard
2.7 方法的回收率及精密度试验
选用汾酒、竹叶青酒共2种基质,分别进行3个浓度水平添加回收试验,每个添加浓度水平做10个平行样品。方法的回收率和精密度(用相对标准偏差(RSD)表示)数据见表1。竹叶青酒样品和加标样品的气相色谱图分别见图2和图3。
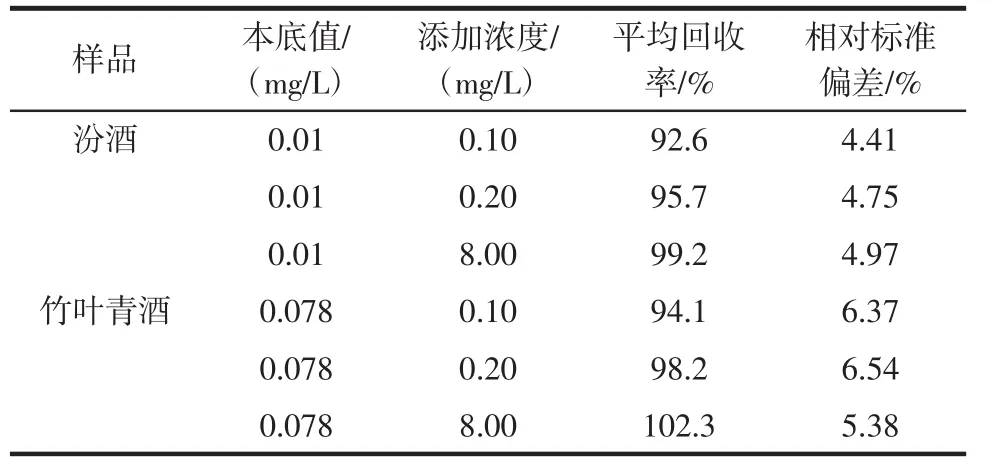
表1 氰离子在不同样品中的回收率和相对标准偏差(n=10)Table 1 Results of recovery and precision tests of cyanide in detection samples(n=10)
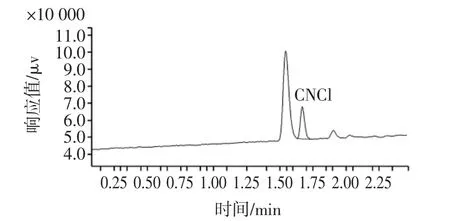
图2 竹叶青酒色谱图Fig.2 Chromatogram of the zhuyeqing
3 结论
本方法直接取酒样品加入衍生剂后,采用顶空气相色谱法检测酒中氰化物。该方法简便、快捷,大大缩短了检测周期,同时对环境没有什么破坏,是一种环保、经济、快速的检测方法。为酒制品的质量保证提供了强有力的支持,尤其是面对大批量样品的检测,更能体现出方法的优越性。
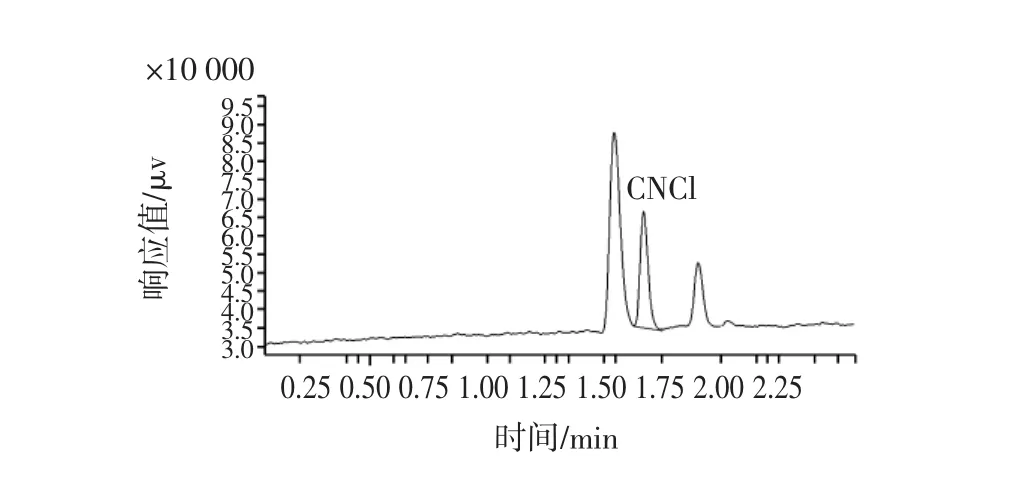
图3 竹叶青酒添加0.1 mg/L氰离子的色谱图Fig.3 Chromatogram of the zhuyeqing with cyanide standard
[1] 李源栋,樊林,钱海燕,等.酒中氰化物测定方法的研究进展[J].酿酒,2009,36(6):16-19
[2] 李美兰.白酒中氰化物的测定方法研究[J].中国卫生产业,2011(4):29
[3]中华人民共和国卫生部.GB 2757-2012食品安全国家标准蒸馏酒及其配制酒[S].北京:中国标准出版社,2012
[4] 周庆龙,王绍杰,张文德.高档名酒中微量氰化物的测定方法[J].中国卫生检验杂志,2005(8):950-969
[5] 吴佑琼.双波长叠加分光光度法测定水中微量氰化物[J].化学分析计量,2011(4):60-62
[6]朱晓玲.水中微量氰化物的异烟酸吡唑啉酮分光光度测定法进展[J].北方环境,2011,23(4):140,157
[7] 崔战友,钟其顶,李国辉,等.酒醅中氰化物的测定方法研究[J].酿酒科技,2015(5):101-103,106
[8] 顾建华,何鹏妍.异烟酸-巴比妥酸分光光度法测定蒸馏酒及其配制酒中氰化物含量[J].中国卫生检验杂志,2015(6):813-815
[9]郑艳春.利用亚甲蓝萃取分光光度法测定粮食中微量氰化物[J].黑龙江粮食,2011(1):35-37
[10]马庆华,冯潇,付元欣,等.Hib多糖蛋白结合疫苗中残余氰化物检测方法的建立[J].微生物学免疫学进展,2011(2):1-7
[11]刘少民,徐娟,闫向阳,等.顶空-毛细管气相色谱法测定卷烟主流烟气中的氰化物[J].中国烟草学报,2008(1):1-5
[12]时振强,肖林,刘德辉.顶空气相色谱法测定酒中微量氰化物[J].中国卫生检验杂志,2002(4):433
[13]叶梅,吴文林,郭靓,等.离子色谱-脉冲安培检测法快速测定配制酒中的氰化物[J].食品科学,2016(8):192-195
[14]张建,苏利进,李凯,等.GC-MS定性/HS-20顶空结合GC定量测定酒中痕量氰化物[J].酿酒科技,2014(7):105-108
[15]中华人民共和国卫生部,中国国家标准化管理委员会.GB/T5009.48-2003蒸馏酒与配制酒卫生标准的分析方法[S].北京:中国标准出版社,2003
Determination of Cyanide in Wine by Headspace Gas Chromatography
DU Li-jun,LIU Xiao-lin,GAO Yuan-hui,GUO Li-hua,LIU Hai-feng,LI Fei-fei
(Technology Center for Inspection and Quarantine of Shanxi Entry-exit Inspection and Quarantine Bureau,Taiyuan 030024,Shanxi,China)
A method for determination of cyanide residue in wine by headspace gas chromatography was described.Win added chloramine T to make cyanide derivative as cyanogen chloride,head-space injection,then separated it by the WAX gas chromatographic column,detected by electron capture detector ECD and quantified by external standard.The results showed as follow,the linear range was 0.001 mg/L-0.1 mg/L,the correlation coefficient was 0.998 and the limit of detection(S/N ≥ 10)was 0.05 mg/L.Recoveries were 4.41%-6.54%with RSD (n=10)at the three different standards levels of 0.1,0.2,8.0 mg/L.The method is simple,rapid,accurate,and can be applied in the determination of cyanide in vinic products,and meet requirements of the daily testing.Meanwhile,the dosage of the derivative,headspace holding time and the temperature were optimized.
wine;headspace gas chromatography;cyanide
2016-09-07
10.3969/j.issn.1005-6521.2017.11.029
国家质检总局科研项目(2015IK215)
杜利君(1979—),男(汉),高级工程师,本科,研究方向:食品安全检测。
猜你喜欢
天津化工(2019年6期)2019-12-10
分析化学(2018年12期)2018-01-22
西藏科技(2016年9期)2016-09-26
湖南大学学报(自然科学版)(2016年6期)2016-07-18
湖南大学学报·自然科学版(2016年6期)2016-07-14
中国农村水利水电(2016年4期)2016-03-23
中国有色冶金(2015年1期)2015-03-06
食品工业科技(2014年9期)2014-03-11
表面工程与再制造(2014年2期)2014-02-27
中国烟草学报(2012年3期)2012-04-10
