
钯催化吲哚的去芳香化双芳基化
2017-07-03 14:57贾义霞李英龙刘人荣
浙江工业大学学报 2017年4期
贾义霞,李英龙,刘人荣
(浙江工业大学 化学工程学院,浙江 杭州 310014)
钯催化吲哚的去芳香化双芳基化
贾义霞,李英龙,刘人荣
(浙江工业大学 化学工程学院,浙江 杭州 310014)
以钯为催化剂,三环己基膦为配体,芳基硼酸为芳基化试剂,通过吲哚的分子内Heck/Suzuki串联反应实现了吲哚的双官能团化,为合成结构多样性的吲哚啉衍生物提供了一种高效、快捷的方法.通过简单起始原料出发,高产率、高对映选择性地合成了一系列的含有连续叔碳/季碳中心的吲哚林化合物.并系统地研究了反应的溶剂、碱和配体对反应的影响以及底物中各种给电子或吸电子基团对产物收率的影响.重要的是,通过该反应可以顺利实现克级规模制备,体现了方法的优越性及可靠性.
钯催化;吲哚;芳基化;去芳构化;串联反应
吲哚啉广泛存在于具有生物活性天然产物以及药物活性分子中,并常用作医药或农药合成的中间体,近年来一直受到化学家们的广泛关注[1-4].大多数已报道的合成吲哚啉方法往往需要多步合成才能得以实现,发展高效、快速构建吲哚啉的合成方法具有重要意义[5].近年来,过渡金属催化的吲哚去芳构化反应已经成为合成具有吲哚啉骨架类化合物的一种重要方法[6].2011年,Bedford小组利用钯催化的偶联反应实现了2-氨基-1,3-二甲基吲哚的去芳构化[7].2012年,You小组报道了钯催化的3-取代吲哚的去芳构化反应,以最高93%的收率获得去芳构化产物[8].2015年,笔者课题组报道了Pd催化的N-取代吲哚的不对称去芳构化反应[9],基于受阻的碳-氢活化的策略首次实现了吲哚的去芳构化还原Heck反应,合成了具有季碳手性中心的吲哚啉类化合物.随后,Lautens课题组以氰化锌为氰基化试剂,首次实现了吲哚2,3-位的去芳构化双官能化反应[10].梁永民小组[11]和笔者小组[12]也分别通过脱羧偶联和sonogashira偶联反应对苄基钯中间体进行捕获,实现了吲哚2,3-位的去芳构化/炔基化反应.最近,Lautens小组又进一步采用芳基硼酸酯为试剂对反应中形成的苄基钯中间体进行捕获,高非对映选择性地完成了吲哚去芳基化/Suzuki偶联串联反应[13].在Lautens课题组进行研究的同时,笔者也对吲哚的2,3-位去芳构化双官能化反应进行研究,与Lautens课题组不同的是笔者采用更便宜的芳基硼酸为底物,以及更简单的醋酸钯/三环己基膦为催化剂,通过吲哚的分子内去Heck/Suzuki串联反应高非对映选择性地实现了吲哚的双官能团化.
1 实验部分
1.1 实验试剂及仪器
仪器:Bruker Advance Ш 500 MHz型核磁共振仪(TMS作为内标,CDCl3作为溶剂)柱色谱为柱层析硅胶(用200~300目硅胶,青岛海洋化工),所有反应均在常规玻璃仪器中进行,采用磁力搅拌.
主要试剂:各类含不同取代基的吲哚、不同取代的邻溴苯甲酸、苯硼酸、醋酸钯、膦配体及各类有机溶剂等(所用试剂均为市售分析纯或者化学纯).
1.2 实验方法
钯催化Heck/Suzuki串联反应对吲哚去芳香化双官能化反应的典型操作步骤:在安培管中依次加入0.2 mmol的取代吲哚底物Ⅰa,0.4 mmol的苯硼酸Ⅱa,0.4 mmol的K2CO3,摩尔分数为5%的Pd(OAc)2和摩尔分数为10%的配体PCy3·HBF4,氮气保护下,注射器加入2 mL的新蒸馏的溶剂,100 ℃下反应12 h.反应结束后粗产品用层析柱分离得到目标产物,其中层析柱分离时使用的淋洗液为V(乙酸乙酯)∶V(石油醚,60~90 ℃)=1∶10.
部分产物表征如下:
10b-甲基-11-(苯基)-10b,11-二氢-6H-吲哚并[2,1-a]吲哚-6-酮(Ⅲaa):白色固体,产率86%,Mp=176~177 ℃,1H NMR(500 MHz, CDCl3)δ7.83(d,J=8.0 Hz, 1H),7.69(d,J=7.5 Hz, 1H),7.41(t,J=7.5 Hz, 1H),7.25(dd,J=12.0,8.5 Hz,2H),7.18(d,J=7.5 Hz,1H),7.13(d,J=7.5 Hz,1H),7.06(d,J=7.5 Hz,1H),6.88~6.96(m,3H),6.55(d,J=7.0 Hz,2H),4.36(s,1H),1.76(s,3H).13C NMR(125 MHz, CDCl3)δ168.5,140.7,139.2,138.9,132.8,131.9,128.7,128.2,128.0,127.6,126.8,126.6,125.0,124.2,123.0,117.1,76.0,57.1,28.4.HRMSm/z(ESI+): Calculated for C22H17NO ([M+H]+):312.138 8, Found 312.138 3.
9-甲氧基-10b-甲基-11-(苯基)-10b,11-二氢-6H-吲哚并[2,1-a]吲哚-6-酮(Ⅲba):白色固体,产率94%,Mp=135-136 ℃,94% yield;1H NMR (500 MHz, CDCl3)δ7.84 (d,J=7.9 Hz,1H),7.42(td,J=7.5,1.0 Hz,1H),7.17~7.20 (m,2H),7.14(td,J=7.0,1.0 Hz,1H),6.94~6.99(m,4H),6.85(dd,J=8.5,2.5 Hz,1H),6.55~6.58(m,2H),4.35(s,1H),3.76(s,3H),1.75(s,3H).13C NMR(125 MHz, CDCl3)δ168.4,159.8,141.1,140.8,139.2,139.0,134.2,128.6,128.2,127.6,126.8,126.6,1 250,123.8,120.4,117.0,106.7,75.6,57.0,55.5,28.5. HRMSm/z(ESI+): Calculated for C23H19NO2([M+H]+):342.149 4, Found 342.148 7.
9,10b-二甲基-11-(苯基)-10b,11-二氢-6H-吲哚并[2,1-a]吲哚-6-酮(Ⅲca):白色固体,产率94%,Mp=172~173 ℃,94% yield;1H NMR(500 MHz, CDCl3)δ7.83(d,J=8.0 Hz,1H),7.58(d,J=7.5 Hz,1H),7.41(td,J=7.5,1.0 Hz,1H),7.17~7.21(m,1H),7.13(td,J=7.5,1.0 Hz,1H),7.05 (dd,J=8.0,0.5 Hz,1H),6.92~6.98 (m,3H),6.86(s,1H),6.55~6.59(m,2H),4.35(s,1H),2.26(s,3H),1.76(s,3H).13C NMR (125 MHz, CDCl3)δ168.7,149.1,142.7,140.8,139.4,138.8,130.3,129.1,128.6,128.1,127.6,126.8,126.5,124.9,124.0,123.6,117.0,75.7,57.1,28.5,21.8. HRMSm/z(ESI+): Calculated for C23H19NO([M+H]+):326.154 5, Found 326.154 2.
9-氟-10b-甲基-11-(苯基)-10b,11-二氢-6H-吲哚并[2,1-a]吲哚-6-酮(Ⅲda):Mp=193~194 ℃,产率68%;1H NMR (500 MHz, CDCl3)δ7.84(d,J=8.0 Hz,1H),7.42~7.46(m,1H),7.36(dd,J=7.5,2.5 Hz,1H),7.21(dd,J=7.0,0.5 Hz,1H),7.15~7.18(m,1H),7.05(dd,J=8.0,4.5 Hz,1H),6.95~7.01(m,4H),6.56(dd,J=7.5,1.5 Hz,2H),4.37(s,1H),1.77(s,3H).13C NMR(125 MHz,CDCl3)δ167.2,167.1,162.6(d,J=246.3 Hz),144.2(d,J=2.4 Hz),140.5,138.9(d,J=19.8 Hz),135.0(d,J=8.4 Hz),128.8,128.4,127.5,127.0,126.6,125.3,124.5(d,J=8.4 Hz),119.5(d,J=24.0 Hz),117.1,110.8(d,J=25.0 Hz),75.7,57.1,28.4. HRMSm/z(ESI+): Calculated for C22H16FNO([M+H]+):330.129 4, Found 330.129 0.
10,10b-二甲基-11-(苯基)-10b,11-二氢-6H-吲哚并[2,1-a]吲哚-6-酮(Ⅲea):Mp=180~181 ℃,产率49%;1H NMR(500 MHz, CDCl3)δ7.86(d,J=7.5 Hz,1H),7.66(d,J=7.5 Hz,1H),7.39(td,J=7.5,1.0 Hz,1H),7.23(t,J=7.5 Hz,1H),7.16(d,J=7.5 Hz,1H),7.08~7.11(m,2H),6.94~6.97(m,3H),6.64~6.67(m,2H),4.39(s,1H),2.27(s,3H),1.81(s,3H).13C NMR(125 MHz, CDCl3)δ167.7,146.3,140.9,140.0,138.0,134.0,133.7,132.5,128.6,128.5,128.0,127.3,126.9,126.3,124.9,122.0,117.0,76.2,56.7,26.5,19.1. HRMSm/z(ESI+): Calculated for C23H19NO([M+H]+):326.154 5, Found 326.154 0.
8,9-二甲氧基-10b-甲基-11-(苯基)-10b,11-二氢-6H-吲哚并[2,1-a]吲哚-6-酮(Ⅲfa):Mp=180~181 ℃,产率96%;1H NMR(500 MHz, CDCl3)δ7.80(d,J=8.0 Hz,1H),7.40(t,J=7.5 Hz,1H),7.14~7.18(m,2H),7.12(d,J=7.5 Hz,1H),6.97(d,J=7.0 Hz,3H),6.59(d,J=6.5 Hz,2H),6.44(s,1H),4.34(s,1H),3.85(s,3H),3.77(s,3H),1.76(s,3H).13C NMR(125 MHz, CDCl3)δ169.1,152.8,149.5,143.1,141.0,139.6,138.5,128.6,128.1,127.8,126.9,126.5,124.8,124.7,116.8 105.4,105.2,75.4,57.07(s),56.1,56.0,28.7. HRMSm/z(ESI+): Calculated for C24H21NO3([M+H]+):372.160 0, Found 372.162 1.
2 结果与讨论
2.1 反应条件优化
采用吲哚衍生物Ⅰa和苯硼酸Ⅱa作为模型底物,来研究吲哚与苯硼酸的去芳构化串联反应.以摩尔分数为5%的Pd(OAc)2作为催化剂,摩尔分数为10%的PPh3作为配体,2当量的K2CO3作为碱,在甲苯溶剂中100 ℃下进行12 h反应后,能够以66%的收率得到单一构型的去芳构化目标产物Ⅲa(表1,序号1).受此鼓舞,首先对反应条件中的溶剂进行筛选,其反应式为

表1 溶剂对产率的影响

序号12345溶剂TolueneDMFTHFMeOHMeCN产率/%664353—78
2.1.1 溶剂对产率的影响
当以DMF和THF作为反应溶剂时,虽然反应也能顺利发生,但产率有所降低(表1,序号2,3).而以MeOH作为溶剂时,反应几乎不能检测到产物(表1,序号4).最后,当使用MeCN作为溶剂时,反应产率可以得到较大提升,以78%收率得到目标产物.
2.1.2 碱对产率的影响
接下来,以MeCN为溶剂对反应的碱进行考察.如表2所示,以K3PO4为碱时能够得到与K2CO3几乎相当的结果.采用有机碱Et3N以及碱性更弱的NaHCO3为碱时,反应产率显著降低.而采用更强碱性的Cs2CO3为碱时,反应几乎得不到产物.
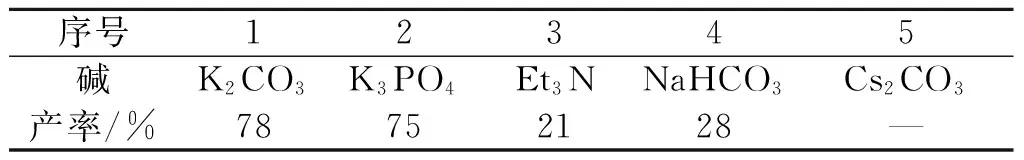
表2 碱对产率的影响
2.1.3 配体对产率的影响
以MeCN为溶剂,K2CO3作碱,对反应的配体进行考察.如表3所示,当使用双齿配体dppm或dppb作为配体时,产率有所下降.而使用大位阻富电子的PCy3为配体时,能以最高86%的收率得到目标产物,而以PtBu3·HBF4为配体时,产率仅有18%.

表3 配体对产率的影响
2.2 底物拓展
在上述最优反应条件下,首先对吲哚底物的普适性进行了考察.底物Ⅰ的拓展结构式(yield表示收率,dr表示非对映异构体比值)为
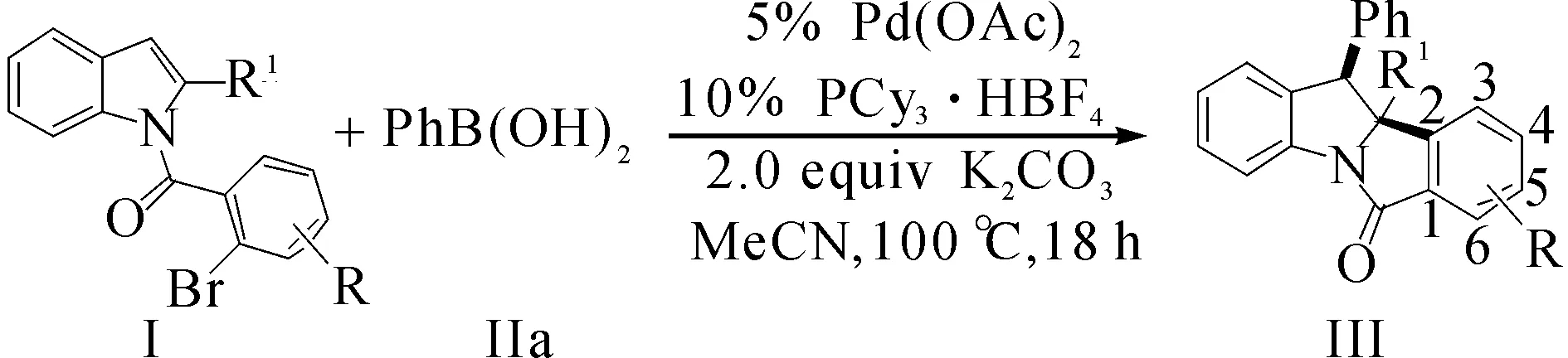
吲哚底物含溴芳环上不论含有富电子还是吸电子取代基,均能以良好到优异的产率得到去芳构化串联反应产物.相对而言,含有给电子基团的底物较吸电子取代基底物产率更高(Ⅲfa vs Ⅲda).当芳环上含有位阻更大的邻位取代基时,反应产率大幅降低(Ⅲea).除此之外,吲哚2位上的取代基为芳香取代基时,同样能以较高收率得到目标产物(Ⅲga~Ⅲia),产率较烷基取代底物略有下降.
接下来,对芳基硼酸底物进行考察.底物2的拓展结构式(yield表示收率,dr表示非对映异构体比值)为


在标准条件下,各种邻位、对位和间位取代的芳基硼酸均能以良好到优秀的收率得到去芳构化目标产物.含有强给电子(Ⅱc)或强吸电子基团硼酸(Ⅱd)都使反应产率有较大幅度下降.当以对氯硼酸作为底物时,反应的非对映选择性较差,只能得到非对映选择性为6∶1的去芳香化产物Ⅲea.总体上讲,含有邻位取代基的芳基硼酸较相应间位或对位产率有较大幅度降低(Ⅲah vs Ⅲai)和(Ⅲaj vs Ⅲab and Ⅲag).
3 结 论
以醋酸钯钯为催化剂,芳基硼酸为芳基化试剂,通过吲哚的分子内去芳构化/芳基化串联反应实现了吲哚的双官能团化,高非对映选择性地合成了一系列含有连续季碳和叔碳中心的2,3-二取代吲哚啉化合物.该反应条件温和、底物适用范围较好.
[1] 沈晓燕,高建荣,韩亮,等.MCM-41的磺酸官能化及催化性能研究[J].浙江工业大学学报,2011,39(6):605-608.
[2] 刘运奎,江波,娄绍杰,等.钯催化的喹喔啉基导向的邻位碳-氢键乙酸化反应[J].浙江工业大学学报,2013,41(1):1-5.
[3] 高建荣,龚渊,金红卫,等.一种合成氮丙啶衍生物的新方法[J].浙江工业大学学报,2014,42(1):77-80.
[4] DALPOZZO R, BARTOLI G, BENCIVENNI G. Recent advances in organocatalytic methods for the synthesis of disubstituted 2-and 3-indolinones[J]. Chemical society reviews,2012,41,7247-7290.
[5] IYENGAR R, SCHILDKNEGT K, MORTON M, et al. Revisiting a classic approach to the aspido-sperma alkaloids: an intramolecular schmidt reaction mediated synthesis of (+)-aspi-dospermidine[J]. Journal of organic chemistry,2005,70(26):10645-10652.
[6] PAPE A R, KALIAPPAN K P, KUNDIG E P. Transition-metal-mediated dearomatization re-actions[J]. Chemical reviews,2000,100(8):2917-2940.
[7] BEDFORD R B, BUTTS C P, HADDOW M F, et al. Reactive 4a-alkyl-4aH-carbazoles by catalytic dearomatisation, and their unusual dimerisation and dealkylation reactions[J]. Chemical communications,2009,32(32):4832-4834.
[8] WU K J, DAI L X, YOU S L. Palladium(0)-catalyzed dearomative arylation of indoles: convenient access to spiroindolenine derivatives[J]. Organic letters,2012,14(14):3772-3775.
[9] SHEN C, LIU R R, FAN R J, et al. Enantioselective arylative dearomatization of indoles via Pd-catalyzed intramolecular reductive Heck reactions[J]. Journal of the american chemical society,2015,137(15):4936-4939.
[10] PETRONE D A, YEN A, ZEIDAN N, et al. Dearomative indole bisfunctionalization via a di-astereoselective palladium-catalyzed arylcya-nation[J]. Organic letters,2015,17(19):4838-4841.
[11] CHEN S, WU X X, WANG J, et al. Palladium-catalyzed intramolecular dearomatization of indoles via decarboxylative alkynyl termi-nation[J]. Organic letters,2016,18(16):4016-4019.
[12] LIU R R, XU T F, WANG Y G, et al. Palladium-catalyzed dearomative arylalkynylation of indoles[J]. Chemical communications,2016,52(94):13664-13667.
(责任编辑:陈石平)
Palladium-catalyzed dearomative diarylation of indole
JIA Yixia, LI Yinglong, LIU Renrong
(College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China)
A novel Pd-catalyzed bisfunctionalization of indoles that proceeds via an intramolecular dearomatization/arylation cascade reaction is developed by employing Pd(OAc)2as a catalyst, PCy3as a ligand and phenylboronic acid as an arylative reagent, which provides a straightforward and efficient access to structural diversely indolines. A series of indolines bearing vicinal tertiary and quaternary stereocenters can be obtained in excellent yields and derived from easily available starting materials. A set of solvents, bases and ligands were screened to enhance the yield. The effect of substituents was also examined. Importantly, this protocol was amenable to gram-scale synthesis, showing the practicality of this reaction.
palladium-catalyzed; indole; arylation; dearomatization; cascade reaction
2016-11-01
国家自然科学基金资助项目(21372202)
贾义霞(1976—),男,江苏泰州人,教授,博士,研究方向为不对称合成,E-mail:yxjia@zjut.edu.cn.
O62
A
1006-4303(2017)04-0387-04
猜你喜欢
无机盐工业(2022年7期)2022-07-11
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
探索科学(学术版)(2021年2期)2021-04-22
核科学与工程(2021年1期)2021-03-05
昆明医科大学学报(2020年12期)2021-01-26
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
科技资讯(2018年16期)2018-10-26
