
Al-Zr系金属间化合物的第一性原理研究
2017-06-29 12:02阮海光黄福祥钟明君陈志谦张照超
重庆理工大学学报(自然科学) 2017年5期
阮海光,黄福祥,钟明君,陈志谦,张照超
(1.重庆理工大学 材料科学与工程学院,重庆 400054;2.西南大学 材料与能源学部,重庆 400715)
Al-Zr系金属间化合物的第一性原理研究
阮海光1,黄福祥1,钟明君1,陈志谦2,张照超1
(1.重庆理工大学 材料科学与工程学院,重庆 400054;2.西南大学 材料与能源学部,重庆 400715)
利用基于密度泛函理论的第一性原理赝势平面波法,计算了Al-Zr系3种金属间化合物在0 K时的生产焓、结合能及相关弹性性能,表征了Al3Zr、Al2Zr和AlZr 3种化合物的结构稳定性、硬度和韧/脆性等,并结合总态密度和分波态密度等电子结构分析,揭示了化合物韧/脆性机制。研究表明:Al3Zr、Al2Zr和AlZr 3种化合物的结构均具有稳定性,Al2Zr在所计算的化合物中具有最高硬度,Al3Zr次之,AlZr最低,且所有化合物均表现为脆性。结合电子结构发现,3种化合物的Al的3s、3p轨道和Zr的4d轨道的价电子具有强烈的杂化作用,从而形成共价键,并导致材料具有低温脆性。
第一性原理;铝锆化合物;稳定性;弹性性能;电子结构
铝合金由于具有优良的综合性能,如比强度和比刚度高、良好的导电性能、耐腐蚀性好,易回收等,目前被广泛用于航天、汽车、电子、生物医学、建筑等领域,并已发展成为继钢铁之后使用最广泛的有色金属材料[1-5]。Zr元素添加到铝合金中始于1956年前苏联 Frindlyander对Al-Zn-Mg-Cu系合金的研究,并引起了其他研究者们的广泛关注[6],其后将Zr作为微量元素添加到 Al-Cu[7]、Al-Li、Al-Cu-Li、A1-Zn-Mg 等系合金中。研究发现,微量元素Zr能抑制合金再结晶,显著提高再结晶温度,并起到细化晶粒尺寸、提高合金强度、改善合金的断裂韧性及抗应力腐蚀性能的作用[6,8-9]。结合Al-Zr二元相图[10]可以发现:Zr添加到铝合金中在常温下基本不固溶在Al中,而是以Al-Zr金属间化合物的形式存在。杨守杰等[11]在研究Zr对超高强铝合金性能影响时,也发现合金中有Al3Zr相产生,能使合金中的位错密度增高,抑制亚晶界的移动,进而提高铝合金强度。而基于第一性原理,Colinet等[12]对不同结构的Al3Zr化合物的稳定性进行了比较,发现 D023>D022>L12。
Zr在铝合金中的作用主要是以金属间化合物的形式体现,此外第一性原理作为有效预测材料稳定性、力学和化学性能等的理论方法,已广泛运用于材料的模拟计算。因此,本文采用第一性原理,对Al3Zr、Al2Zr和AlZr 3种Al-Zr系金属间化合物的热力学稳定性、弹性性能进行计算,并对其韧/脆性行为进行判定,然后利用电子结构对其韧/脆性行为规律进行揭示,进而为铝合金设计提供理论指导,希望能进一步拓展铝合金的应用范围。
1 计算方法与模型
本文采用基于密度泛函理论(DFT)[13]赝势平面波法的CASTEP(cambridge serial total energy package)总能计算软件包[14]对Al-Zr中间化合物进行第一性原理计算。计算过程中选用广义梯度近似(GGA)中的Perdew-Burke-Emzerhof(PBE)势函[15]对化合物体系中的电子与电子间的互换关联能进行处理,并通过自洽迭代法(SCF)对总能进行收敛性计算,收敛差值为5×10-7eV/u。计算时的价电子为Al 3s2 3p1和Zr 4s2 4p6 4d2 5s2。为了提高计算速度和计算精度,所有的计算均执行于倒易空间中,并通过超软(Ultrasoft)赝势平面波动能截断点来控制。经过收敛性测试后,确定布里渊区k点取样为0.35 eV/nm,平面波截断能设置为330 eV。在进行所有计算前,利用Broyden-Flecher-Goldfarb-Shanno(BFGS)[16]方法对化合物晶体结构进行几何优化,以寻找最稳定结构,晶体结构收敛参数如下:体系总能量的收敛精度取5×10-6eV/u,原子平均受力低于0.1 eV/nm,公差偏移小于5×10-5nm ,最大应力偏差值为0.02 GPa。计算所用晶体结构模型如图1所示。
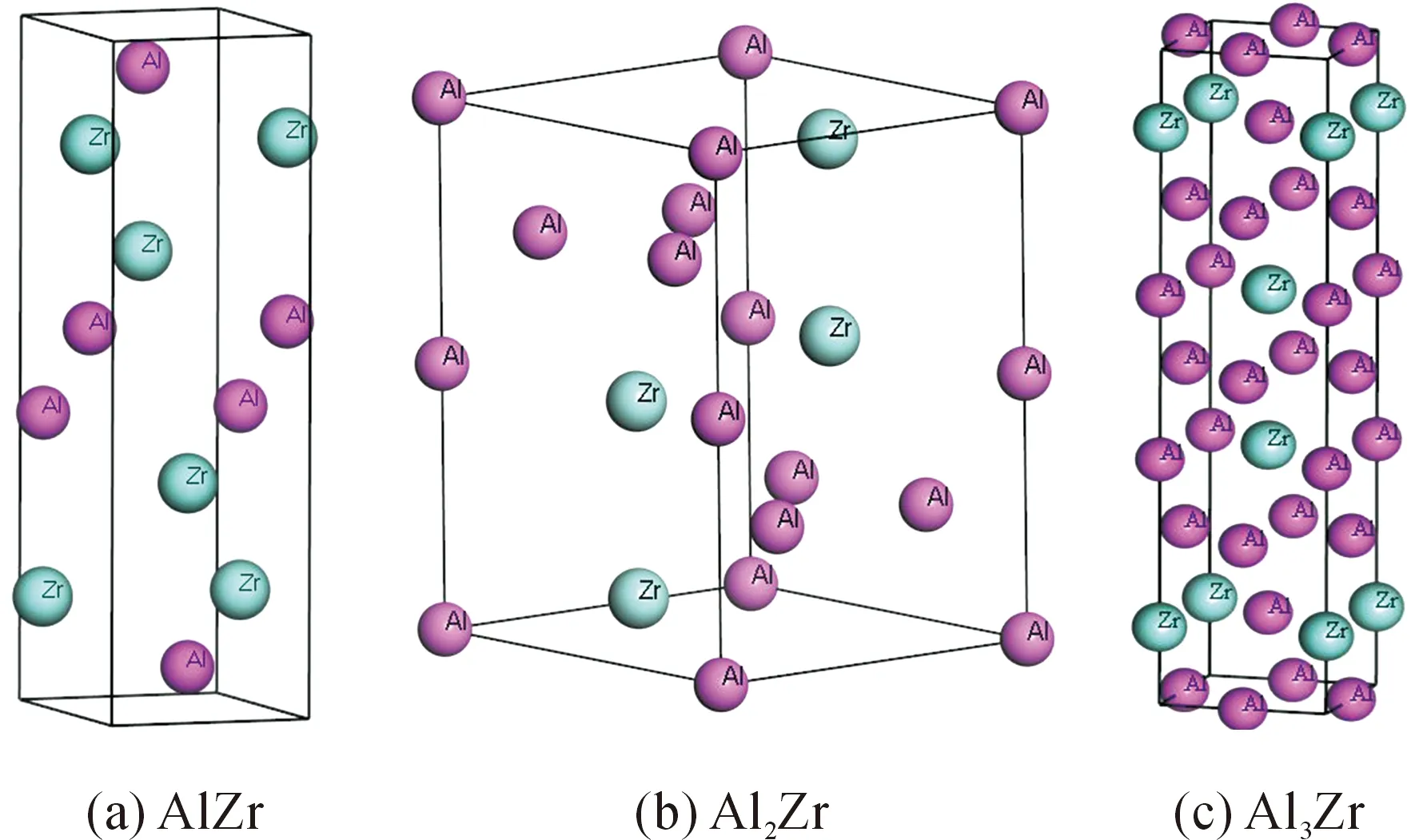
图1 Al-Zr系金属间化合物晶体结构
2 计算结果及讨论
2.1 晶体结构
在本文中,最初的晶体结构建立在Al、Zr和3种Al-Zr系金属间化合物的实验晶体数据基础上,之后利用第一原理计算对化合物的晶格参数和内部坐标进行优化,优化后的晶格参数如表1所示。所有计算值相对实验值偏大,所有相对误差控制在2%以内,且平均偏差大约是1%左右,这可能与计算时所用的关联互换函GGA和忽略了温度影响(计算温度为0 K)有关。总体上Al、Zr和3种Al-Zr系金属间化合物晶格参数的计算值和实验数据基本吻合。表2列出了详细的原子坐标是计算值和其测量值,与测量值相比,平均计算相对误差(1%左右)基本在控制范围内。综上,根据优化后的晶格参数和原子坐标与实验值相比具有一致性,可以确认本文所使用的计算方法和参数设置是合理、可靠的。
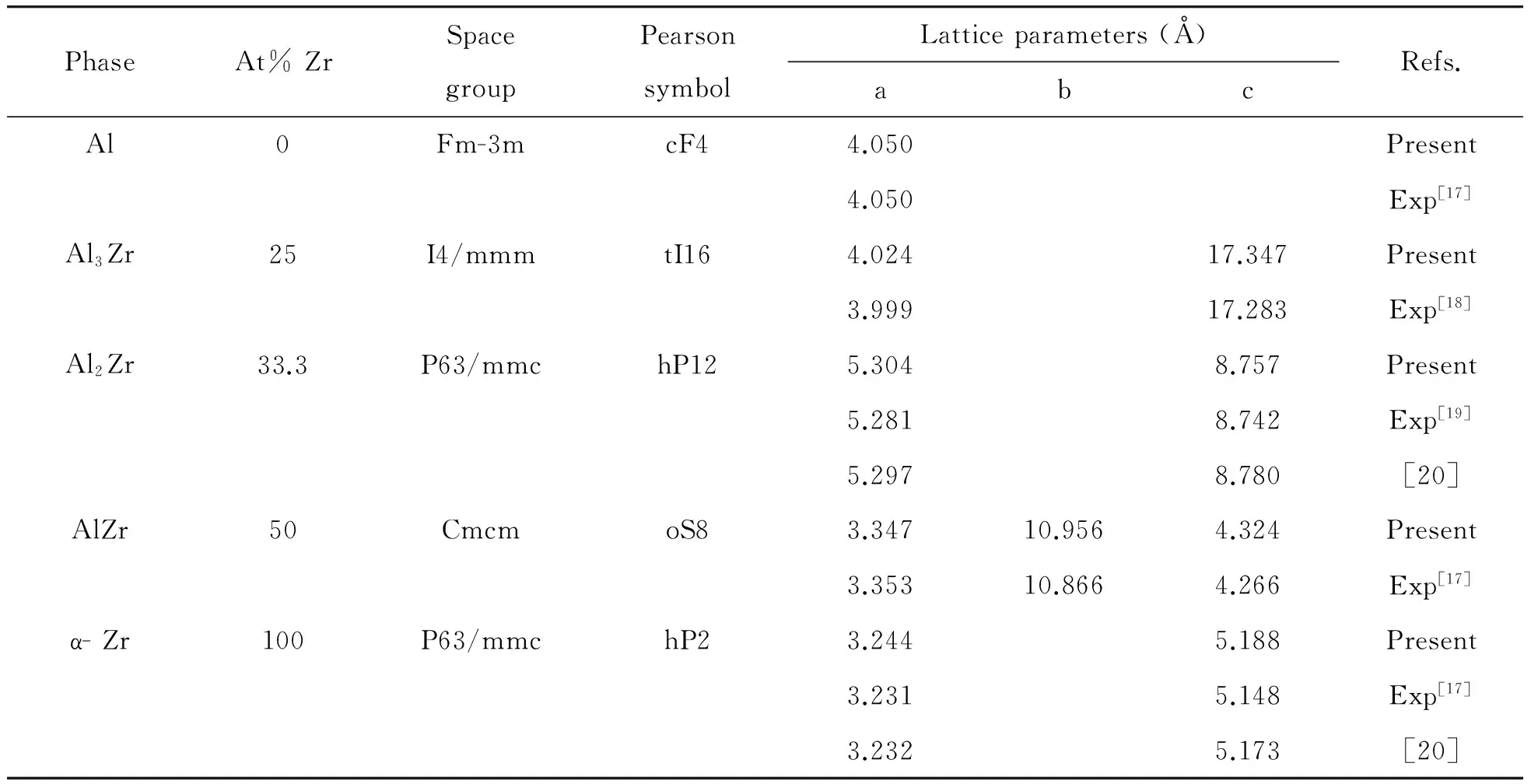
表1 Al、Zr和3种Al-Zr系金属间化合物的晶体参数的计算值和实验值

表2 Al-Zr系金属间化合物的晶体参数(Wyckoff位置)的计算值和实验值
2.2 生成焓及稳定性
处于平衡结构的合金的热力学性能通过其生成焓和结合能进行判定,为了进一步了解Al-Zr系金属间化合物的热力学性能,本文计算了Al3Zr、Al2Zr和AlZr化合物的生成焓和结合能,并对它们的结构稳定性进行了判定。
生成焓是指物质反应后吸收或者释放的能量,当其吸收热量时表现为正值,反之,为负值。它可以用来反映金属间化合物形成的难易程度,当其为负值且绝对值越大时,表明该金属间化合物越容易形成。AlxZry化合物的生成焓(ΔHf)能通过以下公式进行计算:
(1)

晶体的结合能关系到晶体的强度及结构的稳定性,是自由原子结合成晶体时放出的能量。结合能为负值时且绝对值越大,表明晶体结构越稳定。AlxZry化合物的结合能(Ecoh)的计算公式为:
(2)
其中:E(AlxZry)为AlxZry化合物的晶胞总能,E(Al)、E(Zr)分别为独立状态下Al、Zr原子的总能,x,y分别为化学计量式AlxZry中Al、Zr原子数量。
Al-Zr系3种金属间化合物Al3Zr、Al2Zr和AlZr的总能、生成焓和结合能如表3所示。本文所计算的化合物的生成焓和结合能与可利用的其他计算值和实验值[21-23]相比均相符,表明本文所选择的计算方法和参数具有合理性。比较表3中化合物的生成焓可以发现:Al2Zr化合物的生成焓为负值且绝对值最大,故其最容易形成。根据所计算化合物的结合能的绝对值大小顺序:AlZr>Al2Zr>Al3Zr,可以发现所计算的Al-Zr系3种金属间化合物结构稳定性顺序为:AlZr>Al2Zr>Al3Zr,这与Du等[24]得出的结论相一致:在Al-Zr系化合物中随着Zr含量的增大,其结构稳定性增强。此外,也进一步表明随着Zr含量的增大,Al、Zr元素之间化合作用会有所增强。
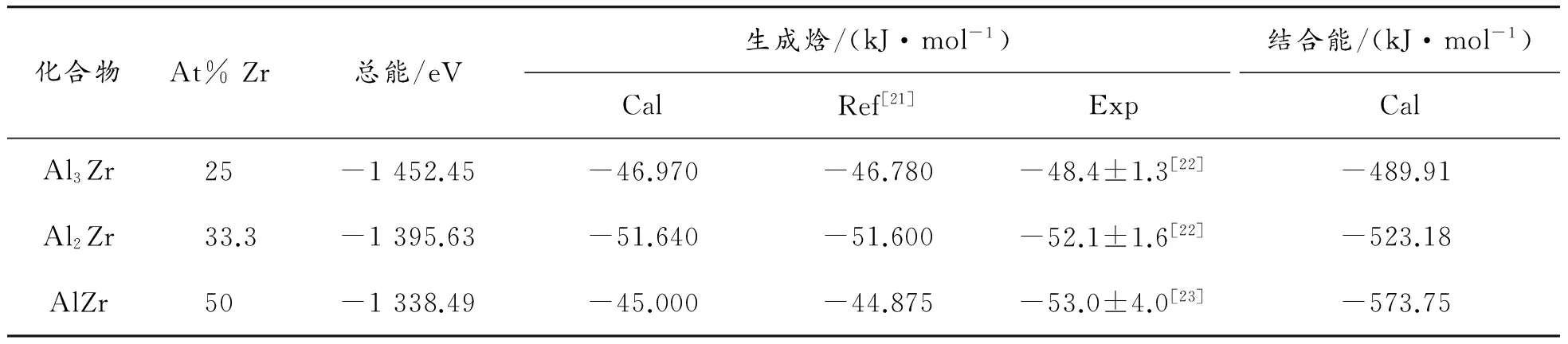
表3 Al3Zr、Al2Zr和AlZr化合物的总能、生成焓和结合能的计算值和其他文献数据
2.3 弹性性能
本文通过给单晶体施加一个线性弹性应变,从而使单晶体产生一被认为是均匀增加的应变能,它们具有以下关系:dE=σijdδij,σij和δij分别为应力和应变张量。晶体的弹性常数是晶体弹性行为最重要的参数,它的一般表达形式为:
(3)
为了研究Al-Zr系金属间化合物的力学行为,分别计算了Al3Zr、Al2Zr和AlZr的弹性常数,其计算值和其他文献数据如表4所示,本文中Al3Zr、Al2Zr和AlZr的弹性常数与Duan等[20]的计算结果相符合。根据Born-Huang的力学稳定性理论,对不同的晶体结构,其稳定性判据是不同的。正方晶系Al3Zr,由于其具有相对较高的对称性,仅有6个独立的弹性常数,其保持稳定性条件为[25]:
(4)
六方晶系Al2Zr,同样有6个独立的弹性常数,其保持稳定性条件为[26]:
(5)
斜方晶系AlZr,则有9个独立的弹性常数,其保持稳定性条件为[27]:
(6)
结合表4所计算的各化合物的弹性常数,可以发现:Al3Zr、Al2Zr和AlZr分别满足各自稳定性条件,表明它们均具有稳定结构。
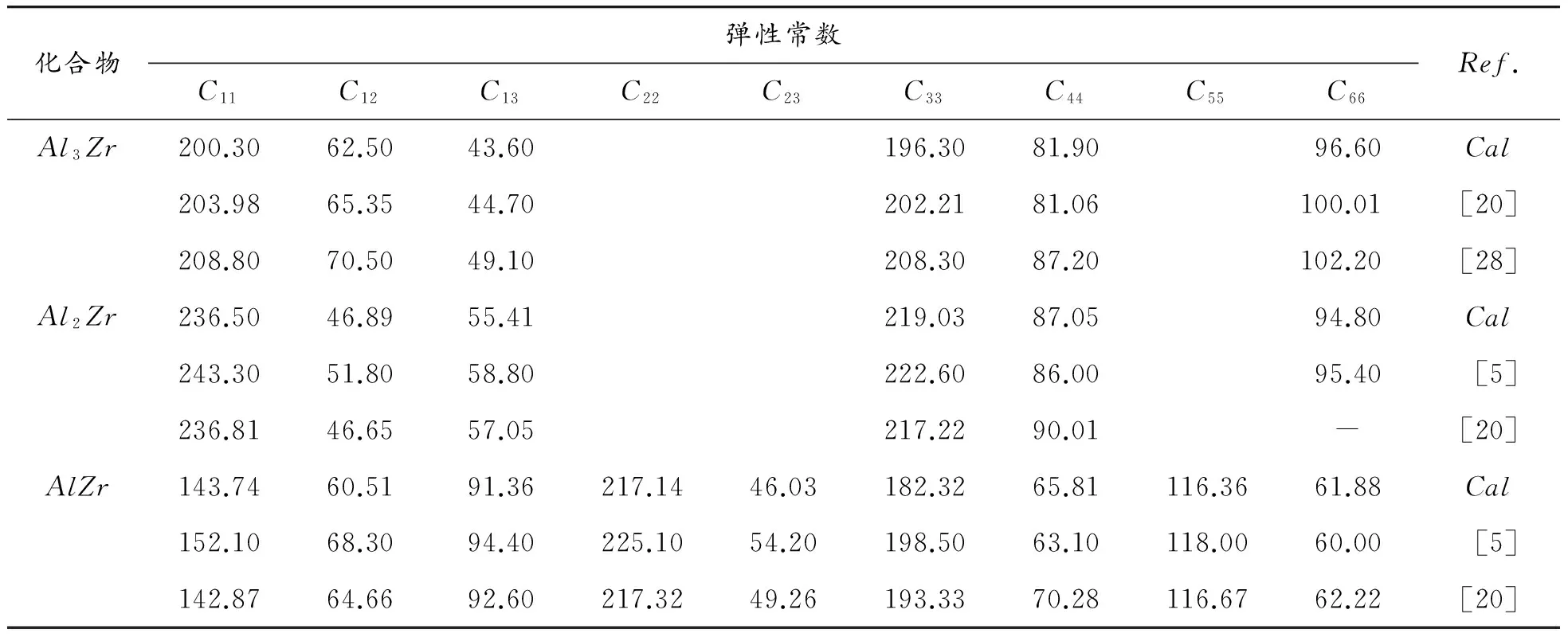
表4 Al-Zr系金属间化合物的弹性常数计算值、实验值和其他计算值
根据单晶材料的弹性常数,结合Voigt-Reuss-Hill(VRH)理论[29],可以进一步计算多晶体材料相关弹性性能,如体模量(B)、剪切模量(G)、杨氏模量(E)、B/G比值、和泊松比(ν),计算公式如下所示:
BV=(C11+C22+C33)/9+2(C12+C13+C23)/9
(7)
GV=(C11+C22+C33)/15-(C12+C13+C23)/15- (C44+C55+C66)/5
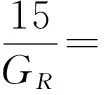
GH=(GV+GR)/2
(8)
公式中Sij为弹性系数,可通过求弹性常数的逆矩阵得到。
(9)
(10)
结合表4及上述计算公式,可以计算出相关弹性性能,如表5所示,计算结果与其他计算值和实验值相符。从表中可以看出:Al2Zr具有最高的体模量(B),AlZr次之,Al3Zr最小。一般来说,材料的体模量的大小与元素的质量密度有关,但是Al-Zr系金属间化合物并不满足这个关系,且体模量作为材料的平均价键强度的评价标准,反映了材料抵抗外力的能力,所以Al2Zr金属间化合物在所计算的化合物中具有最高的抵抗外力的能力。有研究表明:剪切模量(G)、杨氏模量(E)、弹性常数C44在一定程度上能反映材料硬度的大小,并且与之成正相关关系[30]。结合表4、表5可以发现:所计算化合物的剪切模量(G)、杨氏模量(E)、弹性常数C44的大小均按以下顺序排列:Al2Zr、Al3Zr、AlZr,表明Al2Zr在所计算的化合物中具有最高硬度,Al3Zr次之,AlZr最低。此外,杨氏模量(E)也能作为有用的参数来反映材料的刚度大小,杨氏模量(E)越大,表明材料的刚度越大,因此在所计算的Al-Zr系金属间化合物中Al2Zr的刚度最大。
材料的韧/脆性可以根据泊松比(ν)、B/G比值进行判定。泊松比,也叫横向变形系数,是材料在单向受拉或受压时,横向正应变与轴向正应变的绝对值的比值,反映了材料横向变形的难易程度。通常当ν>0.33时,材料表现为塑性,且泊松比越大,表明材料的塑性越好。通过比较可以发现:Al3Zr、Al2Zr、AlZr的泊松比均小于0.33,故可以判定它们均为脆性材料,且Al3Zr的脆性最强,AlZr最弱。此外,B/G比值的大小是判定材料韧/脆性的另外一种方法。Pugh[31]指出当B/G>1.75时,材料表现为塑性;反之,材料表现为脆性,且B/G比值越大,材料的塑性越好。根据表5的计算数据,可以发现Al3Zr、Al2Zr、AlZr的B/G比值均小于1.75,表明它们均为脆性材料,且Al3Zr的脆性最强,AlZr最弱,这与通过泊松比判据得出的结果一致。

表5 Al-Zr系金属间化合物的弹性性能的计算值及其他文献数据
2.4 电子结构
化合物的稳定性及其强度在显微尺度上归因于化合物的电子结构的变化。为了进一步研究Al-Zr系化合物的稳定性和强度变化机理,本文计算了Al3Zr、Al2Zr、AlZr金属间化合物的总态密度(DOS)、分波态密度(PDOS)。
图2是Al-Zr系3种化合物的总态密度(DOS)及分波态密度(PDOS)图。从化合物的总态密度图可以看出:所有化合物的费米能级(0 eV)附近均出现波谷,表明存在赝能隙。态密度曲线以赝能隙为分界线,可以分为成键区和反键区。根据费米能级与赝能隙的位置关系,可以定性地判断化合物结构的稳定性。当费米能级位于赝能隙的右侧,即成键态均被电子所占据,表明化合物具有稳定结构,反之,表明化合物为亚稳定结构[30]。赝能隙的宽度在一定程度上能直接体现化合物共价性的强度,一般来说赝能隙越宽,表明化合物的共价性越强。从图2中可以得出Al3Zr、Al2Zr、AlZr化合物的赝能隙宽度分别为3.68、2.48 eV和2.85 eV,表明Al3Zr的共价性最强,Al2Zr化合物的共价性最弱。本文中所有化合物的费米能级均在赝能隙附近,说明它们都具有较高的稳定结构,这与Al-Zr间存在较强的具有方向性的共价键有关,也进一步说明所计算的Al-Zr系化合物具有低温脆性,这与上述弹性性能的计算结果相一致。此外,Al-Zr系化合物在费米能级处的态密度均不为0,表明Al3Zr、Al2Zr、AlZr都具有金属特性。
如图2(a)所示为Al3Zr金属间化合物的总态密度(DOS)及分波态密度(PDOS)图。由图可见,在费米能级以下的成键电子主要分布在以下3个区域:由Zr的4s和5s轨道的价电子贡献的-49.46~-47.63 eV区间,由Zr的4p轨道的价电子贡献的-27.65~ -25.58 eV区间,由Al的3s、3p轨道和Zr的4d轨道的价电子贡献的-10.51~0 eV区间。 图2(b)、2(c)分别为Al2Zr、AlZr金属间化合物的总态密度(DOS)及分波态密度(PDOS)图,它们和Al3Zr具有类似的成键电子分布。结合分态密度图和总态密度图分析,可以发现Al-Zr系3种化合物的共价性主要归因于Al的3s、3p轨道和Zr的4d轨道的价电子具有强烈的杂化作用。

图2 Al-Zr系化合物的总态密度(DOS)及分态密度(PDOS)图
3 结束语
本文采用基于密度泛函理论(DFT)赝势平面波法的CASTEP总能计算软件包对Al-Zr系3种金属间化合物进行第一性原理计算,研究了Al3Zr、Al2Zr、AlZr化合物的热力学性能,包括生成焓、结合能,结果表明Al2Zr化合物最容易形成AlZr化合物的结构最稳定,且稳定性会随着Zr的原子百分比含量的升高而升高。通过对Al-Zr系3种金属间化合物的单晶体的弹性常数的计算,得到了多晶体材料的体模量(B)、剪切模量(G)、杨氏模量(E)、B/G值和泊松比(ν)。分析发现Al2Zr在所计算的化合物中具有最高硬度,Al3Zr次之,AlZr最低,且Al2Zr的刚度最大。根据泊松比(ν)、B/G值对材料的韧/脆性进行了判定,结果表明:Al3Zr、Al2Zr、AlZr化合物均为脆性材料,且Al3Zr的脆性最强,AlZr最弱。化合物的电子结构分析表明:所计算化合物均具有较高的稳定性和共价性,且Al3Zr的共价性最强,Al2Zr化合物的共价性最弱,3种化合物的共价性主要归因于Al的3s、3p轨道和Zr的4d轨道的价电子具有强烈的杂化作用。
[1] MUTHAIAH V M S,MULA S.Effect of zirconium on thermal stability of nanocrystalline aluminium alloy prepared by mechanical alloying[J].Journal of Alloys and Compounds,2016,688:571-580.
[2] SAHA S,TODOROVA T Z,ZWANZIGER J W.Temperature dependent lattice misfit and coherency of Al3X(X=Sc,Zr,Ti and Nb) particles in an Al matrix[J].Acta Materialia,2015,(89):109-115.
[3] WANG Jiong,SHANG Shun-Li,WANG Yi,et al.First-principles calculations of binary Al compounds:Enthalpies of formation and elastic properties[J].Calphad,2011,35(4):562-573.
[4] WEN Bin,ZHAO Ji-jun,BAI Fu-dong,et al.First-principle studies of Al-Ru intermetallic compounds[J].Intermetallics,2008,16(2):333-339.
[5] 张旭东,王绍青.A13Sc和A13Zr金属间化合物热力学性质的第一性原理计算[J].金属学报,2013 (4):501-505.
[6] 徐向阳.微量Sc、Zr对ZA27合金组织与性能的影响[D].沈阳:沈阳工业大学,2009.
[7] RAGHAVAN V.Al-Cu-Zr (Aluminum-Copper-Zirconium)[J].Journal of Phase Equilibria & Diffusion,2011,32(5):452-454.
[8] LAIK A,BHANUMURTHY K,KALE G B.Intermetallics in the Zr-Al diffusion zone[J].Intermetallics,2004,12(12):69-74.
[9] FULLER C B,SEIDMAN D N,DUNAND D C.Mechanical properties of Al(Sc,Zr) alloys at ambient and elevated temperatures[J].Acta Materialia,2003,51(16):4803-4814.
[10]OKAMOTO H,MASSALSKI T B.Binary alloy phase diagrams requiring further studies[J].Journal of Phase Equilibria,1994,15(5):500-521.
[11]杨守杰,谢优华,陆政,等.Zr对超高强铝合金时效过程的影响[J].中国有色金属学报,2002,12(2):226-230.
[12]COLINET C,PASTUREL A.Phase stability and electronic structure of the HfAl_ {3}compound[J].Journal of Alloys & Compounds,2001,319(1/2):154-161.
[13]KOHN W,SHAM L J.Quantum density oscillations in an inhomogeneous electron gas[J].Physical Review,1965,137(6A):1697-1705.
[14]CLARK S J,SEGALL M D,PICKARD C J,et al.First principles methods using CASTEP[J].Zeitschrift Für Kristallographie,2005,220:567-570.
[15]PERDEW J P,BURKE K,ERNZERHOF M.Erratum:Generalized Gradient Approximation Made Simple[J].Phys.rev.lett,1996,(7):1396.
[16]FISCHER T H,ALMLOF J.General methods for geometry and wave function optimization[J].Journal of Physical Chemistry,1992,96(24):9768-9774.[17]VILLARS P.Pearson’s handbook:crystallographic data for intermetallic phases[M].USA:ASM International,1997.[18]MA Y,R MMING C,LEBECH B,et al.Structure refinement of Al3Zr using single-crystal X-ray diffraction,powder neutron diffraction and CBED[J].Acta Crystallographica,1992,48(1):11-16.
[19]ISRAEL A,JACOB I,SOUBEYROUX J L,et al.Neutron diffraction study of atomic bonding properties in the hydrogen-absorbing Zr(Al x Fe 1-x) 2 system[J].Journal of Alloys & Compounds,1997,253(5):265-267.
[20]DUAN Y H,HUANG B,SUN Y,et al.Stability,elastic properties and electronic structures of the stable Zr-Al intermetallic compounds:A first-principles investigation[J].Journal of Alloys and Compounds,2014,590:50-60.
[21]DAI J H,WU X,SONG Y.Influence of alloying elements on phase stability and elastic properties of aluminum and magnesium studied by first principles[J].Computational Materials Science,2013,(74):86-91.
[22]MESCHEL S V,KLEPPA O J.Standard enthalpies of formation of 4d aluminides by direct synthesis calorimetry[J].Journal of Alloys & Compounds,1993,191(1):111-116.
[23]KEMATICK R J,FRANZEN H F.Thermodynamic study of the zirconium-aluminum system[J].Journal of Solid State Chemistry,1984,54(2):226-234.
[24]DU Jinglian,WEN Bin,MELNIK R,et al.Cluster characteristics and physical properties of binary Al-Zr intermetallic compounds from first principles studies[J].Computational Materials Science,2015(103):170-178.
[25]WALLACE D C,CALLEN H.Thermodynamics of Crystals[J].Eos Transactions American Geophysical Union,1972,80(13):143-143.
[26]KARKI B B,ACKLAND G J,CRAIN J.Elastic instabilities in crystals from ab initio stress-strain relations[J].Journal of Physics Condensed Matter,1997,9(41):8579-8589.
[27]WU Zhijian,ZHAO Erjun,XIANG Hongping,et al.Publisher’s Note:Crystal structures and elastic properties of superhard IrN2and IrN3from first principles[J].Physical Review B,2007,76(5).
[28]NAKAMURA M,KIMURA K.Elastic constants of TiAl3and ZrAl3single crystals[J].Journal of Materials Science,1990,26(8):2208-2214.
[29]HILL R.The Elastic Behaviour of a Crystalline Aggregate[J].Proceedings of the Physical Society,1952,65(5):349-354.
[30]黄豪杰,徐江.Al代位合金化对D88-Ti5Si3力学性能与电子结构影响的第一性原理研究[J].物理化学学报,2015,02:253-260.
[31]PUGH S F.XCII.Relations between the elastic moduli and the plastic properties of polycrystalline pure metals[J].London Edinburgh & Dublin Philosophical Magazine & Journal of Science,2009,45(367):823-843.
[32]曾凡江.纯金属弹性性质及Al-Ce,Nd和Zr二元系统的第一原理研究[D].南宁:广西大学,2007.
(责任编辑 何杰玲)
The First-Principle Study of Al-Zr Intermetallic Compounds
RUAN Hai-guang1, HUANG Fu-xiang1, ZHONG Ming-jun1, CHEN Zhi-qian2, ZHANG Zhao-chao1
(1.College of Materials Science & Engineering, Chongqing University of Technology, Chongqing 400054, China; 2.Faculty of Materials and Energy, Southwest University, Chongqing 400715, China)
Enthalpy, cohesive energy and elastic performance of three kinds of intermetallic compounds in binary Al-Zr compounds at 0 k were calculated with first-principles pseudopotential plane-wave methods based on the functional theory in order to predict the structure stability, hardness and ductile/brittle, etc. Furthermore, the mechanism of the ductile/brittle of compounds was studied with the electronic structure, such as the total density of states and partial wave state density. The results show that the three compounds of Al3Zr, Al2Zr and AlZr have stability structure, and Al2Zr compound has the highest hardness and all compounds are characterized by brittle. Analysis of electronic structure indicated that valence electrons of Al in 3s and 3p orbital and valence electrons of Zr in 4d orbital in three compounds have strong hybrid effect, thus forming a covalent bond between Al-Zr and making materials have low temperature brittleness.
the first-principle; Al-Zr compound; stability; elastic property;electronic structure
2016-12-18 基金项目:重庆市社会民生科技创新专项(cstc2016shmszx80019)
阮海光(1991—),男,硕士,主要从事功能材料及材料计算研究;通讯作者 黄福祥,男,博士,教授,主要从事有色金属功能材料、模具表明强化、机械零件失效分析研究,E-mail:hfx@cqut.edu.cn。
阮海光,黄福祥,钟明君,等.Al-Zr系金属间化合物的第一性原理研究[J].重庆理工大学学报(自然科学),2017(5):60-67.
format:RUAN Hai-guang, HUANG Fu-xiang, ZHONG Ming-jun,et al.The First-Principle Study of Al-Zr Intermetallic Compounds[J].Journal of Chongqing University of Technology(Natural Science),2017(5):60-67.
10.3969/j.issn.1674-8425(z).2017.05.011
O482
A
1674-8425(2017)05-0060-08
猜你喜欢
大学物理(2022年9期)2022-09-28
物理通报(2020年7期)2020-07-01
制造技术与机床(2019年10期)2019-10-26
北京航空航天大学学报(2017年12期)2017-04-23
中南大学学报(自然科学版)(2016年2期)2017-01-19
贵州师范学院学报(2016年3期)2016-12-01
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年3期)2015-11-24
燕山大学学报(2014年6期)2014-03-11
原子与分子物理学报(2014年4期)2014-02-28
