
金华猪和长白猪粪便微生物移植小鼠的肠道古菌结构分析
2017-06-24 10:54肖英平王军军李天天何祥祥李开锋
动物营养学报 2017年6期
肖英平 王军军 李天天 唐 标 何祥祥 李开锋 杨 华*
(1.浙江农业科学院农产品质量标准研究所,杭州310021;2.北京食品营养与人类健康高精尖创新中心,北京100193;3. 中国农业大学动物科技学院,北京100193)
金华猪和长白猪粪便微生物移植小鼠的肠道古菌结构分析
肖英平1王军军2,3李天天2,3唐 标1何祥祥1李开锋1杨 华1*
(1.浙江农业科学院农产品质量标准研究所,杭州310021;2.北京食品营养与人类健康高精尖创新中心,北京100193;3. 中国农业大学动物科技学院,北京100193)
本试验旨在比较肥胖型的金华猪和瘦肉型的长白猪肠道古菌结构的差异,分析菌群移植小鼠肠道古菌结构。采集金华猪和长白猪的新鲜粪便,通过灌胃移植到经广谱抗生素处理过的小鼠肠道中,提取2种猪粪便及其移植小鼠的盲肠内容物总基因组DNA,使用古菌通用引物对古菌16S rRNA的V4区进行PCR扩增,通过Illumina Hiseq测序平台对扩增产物进行高通量测序,使用QIIME等软件对测序序列进行分析统计。结果表明:金华猪和长白猪粪便及其移植小鼠盲肠内容物中的古菌主要包含3个门,分别为奇古菌门(Thaumarchaeota)、广古菌门(Euryarchaeota)和泉古菌门(Crenarchaeota),其中金华猪粪便中广古菌门的丰度高于长白猪;在属水平上主要包含4个已知属,优势属为餐古菌属(Cenarchaeum)。二者移植小鼠的盲肠内容物古菌结构,在门水平上,移植小鼠的奇古菌门比例占70%以上;奇古菌门的古菌几乎全部来源于餐古菌属;在门和属的水平上,金华猪和长白猪粪便移植小鼠的盲肠古菌结构无显著差异(P>0.05),但基于操作分类单元(OTU)水平的主成分分析显示出很好的组内聚集。由此可见,奇古菌门的餐古菌属是金华猪和长白猪粪便和其移植小鼠盲肠中的优势古菌;金华猪粪便中广古菌门的丰度高于长白猪粪便,且二者移植小鼠盲肠内容物之间古菌结构在OTU水平上存在一定的差异。
古菌;金华猪;长白猪;粪菌移植;高通量测序
猪肉历来是我国居民肉类消费品的主要来源,在国计民生中占有重要地位。金华猪是我国著名的地方品种,有“中国熊猫猪”的美誉,其头部和尾部为黑色,故又名两头乌。具有皮薄骨细、肉质鲜美、性成熟早、繁殖能力强、杂交优势显著的特点,但其生长速度慢、瘦肉率低[1]。而长白猪(Landrace pig)是优良的外来品种,原产于丹麦,体躯瘦长,皮肤、被毛呈白色,生长速度快,瘦肉率高,但存在体质较弱、抗逆性较差、对饲养条件要求较高、繁殖能力低下等缺点[2]。
肠道微生物区系是哺乳动物体内最复杂和最大的微生态系统,对宿主的健康、进化及行为产生明显的影响[3-4]。古菌(Archaea)是肠道微生物的重要组成部分之一。古菌最初发现于高热、高盐度、缺氧的极端环境中(如火山口、盐湖),后通过16S rRNA的比较,发现其明显不同于细菌(Bacteria)和真核生物(Eukarya),与后两者并列称为三域系统[5-6]。古菌广泛分布于各种自然环境中,如海洋、热泉、盐碱湖、土壤、沼泽以及动物的消化道。目前广古菌门(Euryarchaeota)和泉古菌门(Crenarchaeota)是研究较多的古菌,尤其是前者,包含了一大类产甲烷菌,广泛存在于各类动物的消化道,是动物营养研究的热点之一[7-9]。肠道古菌也有助于维持消化道中良好的微生态系统。而奇古菌门(Thaumarchaeota)是近年来发现的一个新的古菌类群,广泛存在于中温环境中,可能在自然界重要元素(氮、碳)的生物地球化学循环中发挥着重要作用[10-12],但其在动物消化道中的分布及功能报道很少[13-14]。
近年来,粪菌移植(fecal microbiota transplantation,FMT)作为一种历史悠久且可以重建肠道菌群的疗法重新被临床所关注,为治疗肠道内及肠道外疾病提供帮助[4,15]。与此同时,作为一种功能获得性(gain-of-function)试验手段,FMT也越来越多被应用于动物的微生物结构功能研究[16],小鼠是其中最常用的动物模型之一[17-18]。研究发现,将猪和斑马鱼的肠道微生物移植到小鼠肠道中,移植供体的菌群结构在一定程度被宿主影响和重塑[19-21]。但目前在菌群移植过程中,普遍关注的是细菌结构,而对于古菌在移植状态下的情况研究较少。本试验通过粪菌移植,分别构建了模拟金华猪和长白猪2种猪肠道微生物环境的小鼠模型,采用高通量测序技术分析了2种猪粪便及其移植小鼠盲肠内容物的古菌结构,为研究跨物种粪便微生物群移植过程中古菌结构的变化提供了数据资料。
1 材料与方法
1.1 试验动物
动物试验在北京斯贝福实验动物科技公司进行。选取28日龄断奶的无特定病原菌(SPF)C57BL/6J小鼠24只,雌雄各12只,饲养在无菌隔离包中,饲喂无菌鼠粮。室内温度保持恒温23 ℃,相对湿度为63%,昼夜光照交替时间为12 h/12 h。
1.2 小鼠抗生素处理
试验采取雌雄分养,每个笼子6只小鼠,参照Wang等[22]的方法,采用广谱抗生素处理小鼠以构建无菌小鼠模型。具体方法为:在小鼠饮用水中加入0.5 g/L万古霉素、1 g/L硫酸新霉素、1 g/L甲硝唑和1 g/L氨苄西林,自由口服28 d。
1.3 猪粪便处理
分别采集1头产2胎的经产金华猪母猪和长白猪母猪新鲜粪便,参照Pang等[23]的方法,制备移植供体的肠道微生物。具体方法为:将新鲜粪便迅速悬浮于预还原处理过的无菌生理盐水中(每克粪便加入4 mL生理盐水,即稀释5倍),漩涡混匀1 min,静置 1 min,快速吸取上清,盛于1.5 mL灭菌处理过的离心管中于-20 ℃保存用于菌群移植;同时另取一部分上清用于高通量测序分析古菌结构。
1.4 试验鼠接种和取样
取上述抗生素处理的小鼠,分成2组,每组雌雄各6只,采用灌胃针分别用金华猪和长白猪粪便菌悬液进行灌胃,每次0.2 mL,连续灌注7 d。接种后连续饲养28 d(即肠上皮及其上覆黏液层经过几轮更新后),屠宰小鼠取盲肠内容物用于高通量测序分析古菌结构。选取盲肠是由于其管腔内容物可以简便且可靠地获得,且其中的微生物种群丰富[20]。
1.5 DNA提取和古菌16S rRNA基因扩增
用QIAamp DNA Stool Mini Kit(QIAGEN公司)试剂盒抽提金华猪和长白猪粪便菌悬液及其灌胃小鼠盲肠内容物总基因组DNA,采用古菌通用引物519F(5′-CAGCCGCCGCGGTAA-3′)和Arch915R(5′-GTGCTCCCCCGCCAATTCCT-3′)对古菌16S rRNA基因V4区进行扩增。
1.6 高通量测序分析
测序由北京诺禾致源生物信息科技有限公司完成。采用Illumina Hiseq高通量测序平台对金华猪和长白猪粪便菌悬液及其灌胃小鼠盲肠内容物古菌16S rRNA基因V4区进行测序。
1.7 数据处理与分析
根据条码序列(barcode)将双端测序序列(read)与样品进行匹配,而后切除条码序列及引物序列。使用Flash 1.2.7软件(http://ccb.jhu.edu/software/FLASH/)将双端序列进行融合(merge),获得未加工序列。随后使用QIIME 1.7.0软件(http://qiime.org/scripts/split_libraries_fastq.html)对序列进行质控和过滤,获得优质序列[24-25]。再通过UCHIME算法将其与参考数据库(Gold database,http://drive5.com/uchime/uchime_download.html)进行比对,发现并去除嵌合体序列,从而获得有效序列[26-27]。
使用Uparse 7.0.1001软件(http://drive5.com/uparse/)根据相似性≥97%原则将通过质控的有效序列聚类成为操作分类单元(operational taxonomic unit,OTU)[28]。采用QIIME 1.7.0默认参数计算各样品的alpha多样性指数和物种分布。为了比较2种猪粪便移植的小鼠盲肠内容物的古菌构成的差异,使用R 2.15.3软件中的FactoMineR软件包对样品之间的古菌结构相似度进行主成分分析(PCoA)。使用ggplot 2软件包绘制散点图。
2 结 果
2.1 测序数据及OTU序列信息统计
由表1可见,经过质控、过滤、拼接、去除嵌合体序列等处理,本试验最终获得1 428 899条古菌有效序列,其中金华猪粪便样品有60 979条有效序列,长白猪粪便样品有61 369条有效序列,24例移植小鼠盲肠样品中的古菌有效序列最少为46 362条,最多为61 524条,个体间有效序列数差异性不大。所获得的古菌有效序列根据97%相似性水平进行OTU聚类分析,金华猪粪便样品最终获得307个OTU,长白猪粪便样品最终获得202个OTU,24例移植小鼠盲肠样品最终获得104~133个OTU。各样品的覆盖度指数为0.999~1.000,表明样本中序列没有被测出的概率极低,因此本次测序结果能够代表样本的真实多样性组成。
2.2 alpha多样性分析
根据97%相似性水平下OTU的信息,使用Chao Ⅰ指数、ACE指数、香浓(Shannon)指数及辛普森(Simpson)指数对样本内古菌群落的丰富度和多样性进行评估,结果见表1。OTU数量(123±2 vs. 117±2,P=0.040 5)、Chao Ⅰ指数(130.180±11.084 vs. 122.663±8.538,P=0.076 2)和ACE指数(130.940±10.915 vs. 123.683±7.905,P=0.076 8)比较一致地显示出金华猪粪便移植小鼠盲肠内容物(JM1~12)的古菌群落丰富度略高于长白猪粪便移植小鼠(LM1~12),但在Chao Ⅰ指数和ACE指数上差异不显著(P>0.05)。Shannon指数(3.811±0.065 vs. 3.898±0.063,P=0.345 4)和Simpson指数(0.882±0.007 vs. 0.889±0.006,P=0.507 6)显示,金华猪粪便移植小鼠盲肠内容物(JM1~12)的古菌群落多样性略高于长白猪粪便移植小鼠(LM1~12),但差异不显著(P>0.05)。
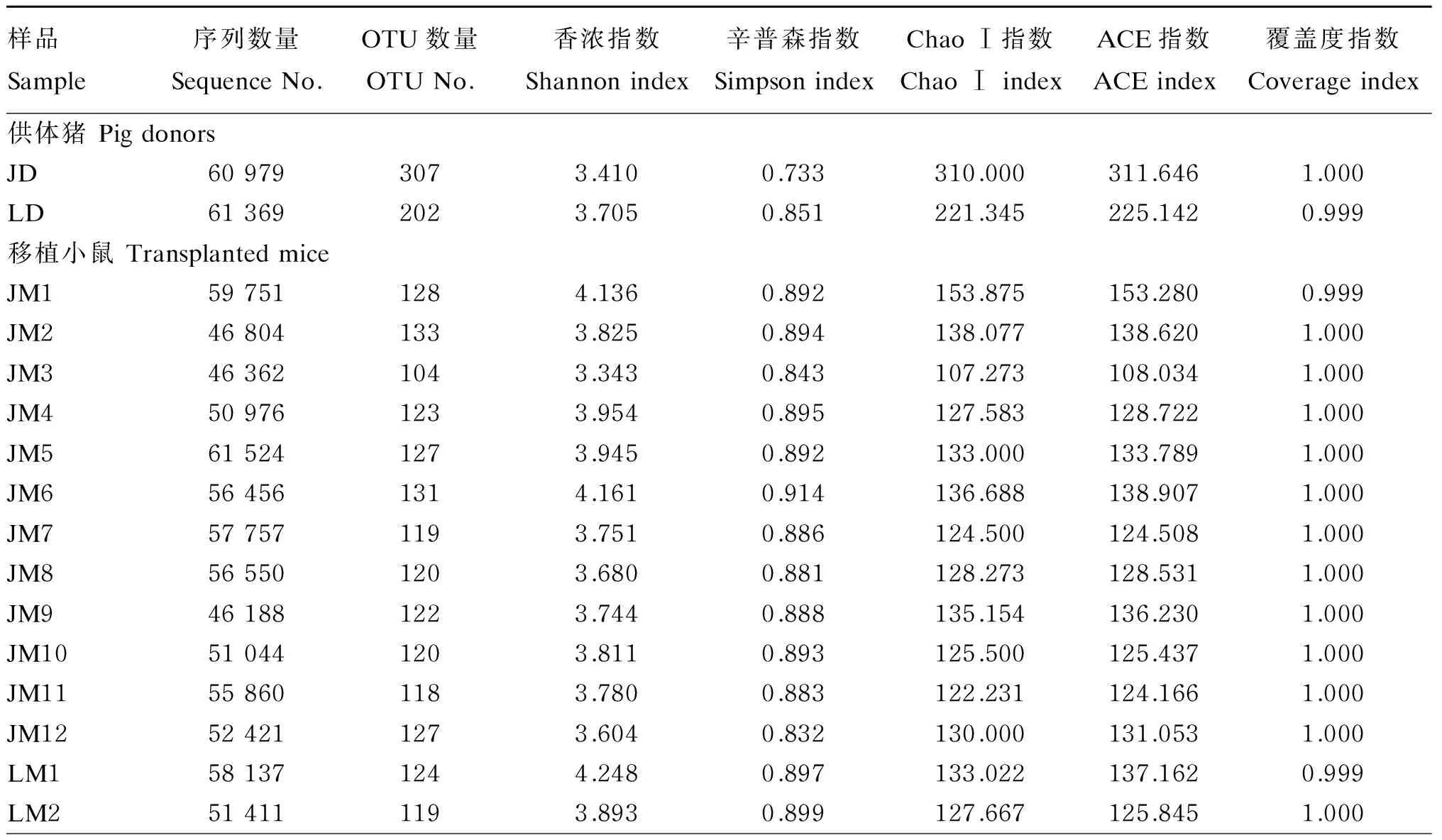
表1 样品测序结果Table 1 Sequencing results of samples

续表1样品Sample序列数量SequenceNo.OTU数量OTUNo.香浓指数Shannonindex辛普森指数SimpsonindexChaoⅠ指数ChaoⅠindexACE指数ACEindex覆盖度指数CoverageindexLM3580751133.8350.900114.750116.4811.000LM4525121133.9020.900117.714120.8681.000LM5535791284.0880.896140.000137.3091.000LM6545161204.0190.882127.333127.3001.000LM7580311113.9990.905116.500117.4481.000LM8591971123.6390.881116.231119.0191.000LM9513281123.6430.879117.000118.3711.000LM10574611224.1950.917128.600128.6551.000LM11559141153.6480.866121.000123.4091.000LM12546971103.6710.842112.143112.3291.000
JD:金华猪粪便样品feces sample ofJinhuapig;LD:长白猪粪便样品 feces sample of Landrace pig;JM:金华猪粪便移植小鼠盲肠内容物cecal contents of mice transplanted withJinhuapig feces;LM:长白猪粪便移植小鼠盲肠内容物cecal contents of mice transplanted with Landrace pig feces。下表同 The same as below。
2.3 移植供体古菌结构
将作为菌群移植供体的金华猪和长白猪粪便样品中获得的OTU分别在门和属水平上进行物种注释,具体结果见表2和图1。可以看出,本试验所得到的供体OTU近1/2都属于奇古菌门(Thaumarchaeota),而广古菌门(Euryarchaeota)和泉古菌门(Crenarchaeota)丰度相对较低。金华猪粪便样品中广古菌门丰度高于长白猪粪便样品。在属水平上,奇古菌门的古菌几乎全部来源于餐古菌属(Cenarchaeum),长白猪粪便样品中还有少量来源于亚硝化球菌属(Nitrososphaera);广古菌门中,金华猪粪便样品中甲烷短杆菌属(Methanobrevibacter)丰度相对较高,此外还有极少量的待定属CandidatusMethanomethylophilus,而长白猪粪便样品中甲烷球菌属(Methanosphaera)丰度较高。

表2 移植供体粪便在不同分类水平上的古菌组成及其丰度Table 2 Archaeal composition and their abundance at different classification levels in feces of transplant donors

A:门水平 phylum level;B:属水平 genus level。
图1 金华猪供体(JD)和长白猪供体(LD)粪便古菌结构
Fig.1 Fecal archaea community structures ofJinhua(JD) and Landrace pig donors (LD)
2.4 移植小鼠盲肠内容物古菌结构
通过灌胃的方式,将金华猪和长白猪粪便微生物分别移植到抗生素处理的无菌小鼠体内,接种后28 d取小鼠盲肠内容物用于古菌结构测序分析。所获得的OTU分别在门和属水平上进行物种注释,具体结果见表3和图2。可以看出,在门水平,移植小鼠的古菌OTU中奇古菌门(Thaumarchaeota)的比例增加到70%以上,而广古细菌门(Euryarchaeota)和泉古细菌门(Crenarchaeota)几乎未检出。而在属水平上,奇古菌门的古菌几乎全部来源于餐古菌属(Cenarchaeum)。2组移植小鼠之间古菌结构在门和属水平上无显著差异(P>0.05),但与供体的古菌结构差别较大。
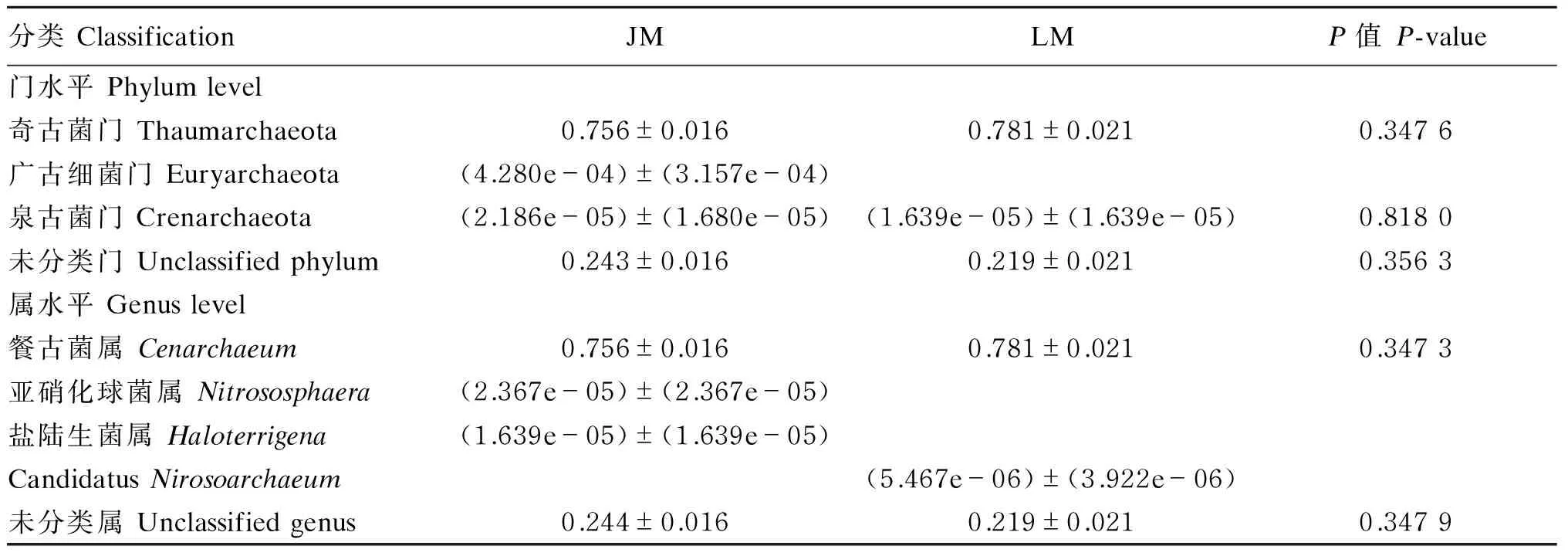
表3 移植小鼠盲肠内容物在不同分类水平上的古菌组成及其丰度Table 3 Archaeal composition and their abundance at different classification levels in cecal contents of transplanted mice
2.5 移植小鼠样品的beta多样性分析
根据各个样品OTU的种类及其丰度计算各个样品间的加权UniFrac距离(weighted UniFrac distance),并基于此,对样品进行了主成分分析(图3)。可以看出,除了JM12和LM12距离较远外,JM组和LM组内其他样品聚集度都较高,且2组在PC2维度上依据其组别显示出明显的组内聚集。
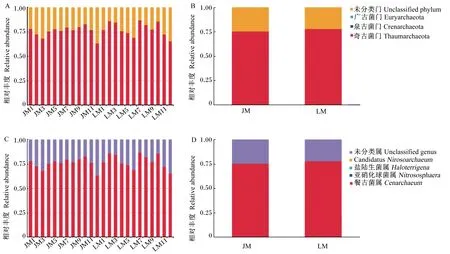
A:移植小鼠门水平的古菌结构 archaea community structure of transplanted mice at phylum level;B:24只移植小鼠门水平的古菌平均值 mean of archaea of 24 transplanted mice at phylum level;C:移植小鼠属水平的古菌结构 archaea community structure of transplanted mice at genus level;D:24只移植小鼠属水平的古菌平均值 mean of archaea of 24 transplanted mice at genus level。
图2 金华猪粪便移植小鼠(JM)和长白猪粪便移植小鼠(LM)盲肠内容物古菌结构
Fig.2 Archaea community structures of cecal contents of transplanted mice using feces formJinhua(JM) and Landrace pigs (LM)
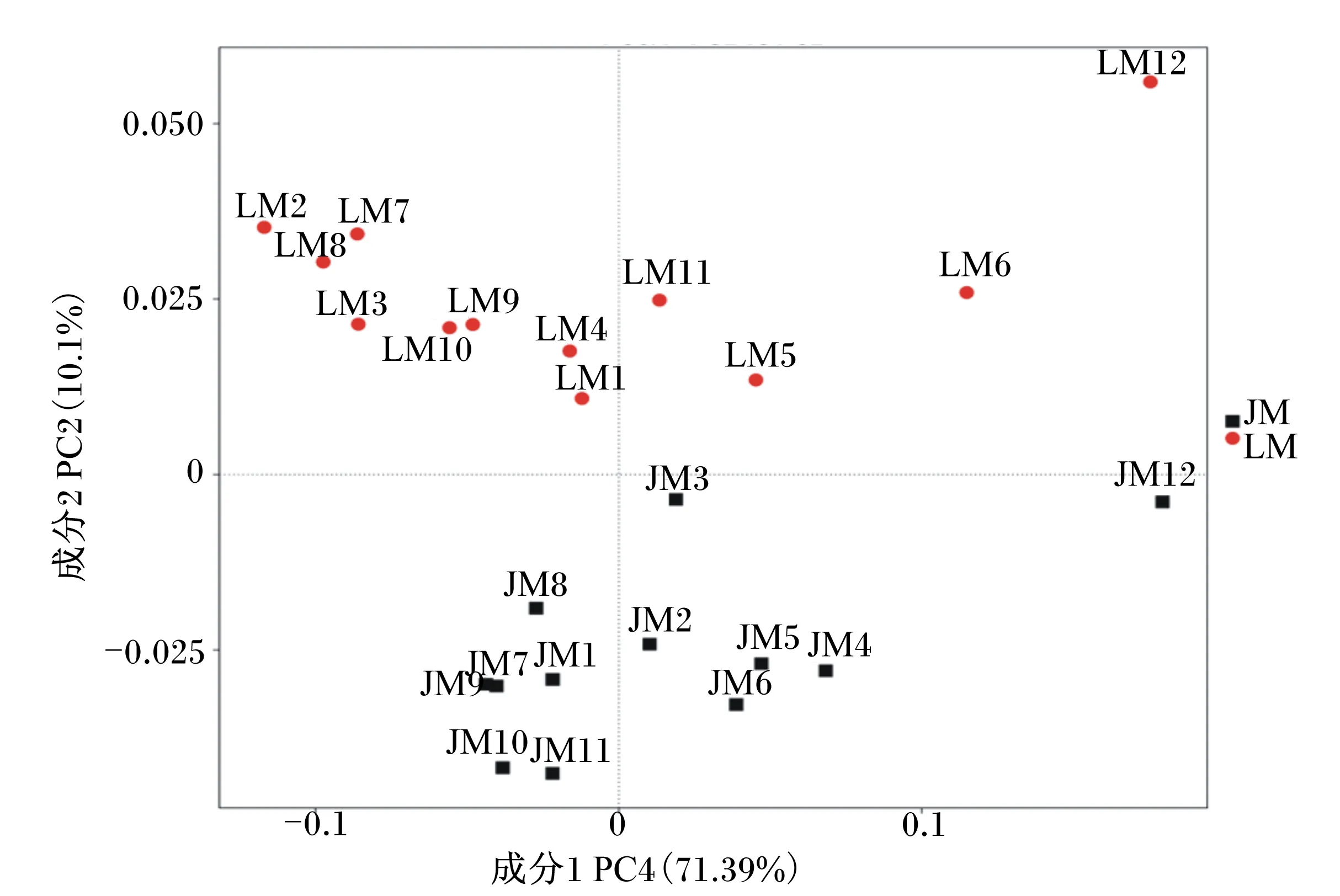
图3 金华猪粪便移植小鼠(JM)和长白猪粪便移植小鼠(LM)古菌结构的主成分分析Fig.3 Principal coordinate analysis of archaea community structures between transplanted mice using feces form Jinhua (JM) and Landrace pigs (LM)
3 讨 论
3.1 金华猪和长白猪粪便古菌结构的异同
金华猪是我国著名的地方品种,生长速度慢,是脂肪型猪,而长白猪是优良的外来品种,生长速度快,是瘦肉型猪[1]。对不同品种猪肠道微生物进行跨物种移植以分析其功能具有重要的研究意义[19],但肠道古菌在此过程中的作用目前研究还较少。本试验通过粪菌移植,分别构建了模拟2种猪肠道微生物环境的小鼠模型,采用高通量测序技术,分析金华猪和长白猪粪便古菌结构差异,并探讨了其粪菌移植小鼠的古菌结构异同,为进一步的功能及机制研究奠定了基础。
以往的研究显示,对动物和人肠道内古菌的研究主要集中在产甲烷菌上。研究表明,动物肠道产甲烷菌的多样性与其生存环境、发育情况及饲料等因素密切相关,因而,不同品种动物肠道内产甲烷菌的结构和组成存在一定差异[7-9]。在脂肪代谢方面,有报道指出,瘦肉型的长白猪粪便中产甲烷古菌的密度和多样性均高于肥胖型二花脸猪,其原因在于甲烷的生成会影响能量获取及体脂肪形成[7];而Zhang等[29]的研究则显示,肥胖个体肠道中利用氢气(H2)的产甲烷古菌含量显著高于瘦的或接受了胃肠旁路手术的个体,同时其产H2细菌含量也较高,细菌和古菌之间跨物种的H2转移被认为是肥胖个体大肠能量摄取增加的重要机制。由此可见,由于研究对象和研究方法的不同,对于动物和人肠道古菌的研究结果还存在一定的差异。而本研究显示,广古菌门虽然在2种猪粪便中含量均不高,但其在金华猪粪便中的丰度明显高于长白猪粪便。其对于2种猪的生长和代谢的影响,还有待进一步的研究。
奇古菌除了在某些白蚁和蚕中[13-14]以外,在人和动物消化道中的分布仅有少量报道,且含量极低[30]。但本研究却发现,在金华猪及长白猪粪便的古菌中,奇古菌门是优势菌,占所有古菌的将近1/2,其中餐古菌为优势菌属。这一结果与以往的研究不一致,其原因及意义还需要进一步试验验证和探讨。
3.2 粪菌移植过程中古菌结构的变化
2006年John Rawls等研究人员将斑马鱼的肠道微生物群到无菌的小鼠肠道,结果发现移植后菌群的构成发生了明显变化,反之亦然。这表明,在跨物种动物间粪便微生物群移植过程中,供体微生物群会被宿主影响和重塑[20]。本试验结果证实,在从猪到小鼠的粪菌移植过程中,古菌结构同样会受到宿主的影响。在本次试验所选取的2个猪品种,金华猪粪便中广古菌门的丰度明显高于长白猪粪便,但移植到小鼠肠道内后这种差异不再明显,且从门到属的水平上,古菌结构均发生了较大的变化,奇古菌门的餐古菌属优势更加明显。虽然在在门和属水平上,金华猪和长白猪粪便移植小鼠肠道古菌结构无明显变化,但在基于OTU水平的主成分分析中,二者有显著不同的聚类,说明古菌结构在OTU水平上存在一定的差异。
4 结 论
① 奇古菌门为金华猪和长白猪粪便古菌组成中的优势菌。
② 广古菌门在金华猪粪便中的丰度明显高于长白猪粪便。
③ 将2种猪的粪菌移植到广谱抗生素处理过的小鼠肠道后,移植小鼠的盲肠内容物古菌结构与供体相比差异明显,同时2种猪粪便微生物移植小鼠的古菌结构在OTU水平上存在一定的差异。
[1] MIAO Z G,WANG L J,XU Z R,et al.Developmental changes of carcass composition,meat quality and organs in theJinhuapig and Landrace[J].Animal,2009,3(3):468-473.
[2] GUO J,SHAN T,WU T,et al.Comparisons of different muscle metabolic enzymes and muscle fiber types inJinhuaand Landrace pigs[J].Journal of Animal Science,2011,89(1):185-191.
[3] HARTSTRA A V,BOUTER K E,BCKHED F,et al.Insights into the role of the microbiome in obesity and type 2 diabetes[J].Diabetes Care,2015,38(1):159-165.
[4] KONTUREK P C,HAZIRI D,BRZOZOWSKI T,et al.Emerging role of fecal microbiota therapy in the treatment of gastrointestinal and extra-gastrointestinal diseases[J].Journal of Physiology and Pharmacology,2015,66(4):483-491.
[5] EME L,DOOLITTLE W F.Archaea[J].Current Biology,2015,25(19):R851-R855.
[6] GRIBALDO S,BROCHIER-ARMANET C.The origin and evolution of Archaea:a state of the art[J].Philosophical Transactions of the Royal Society of London.Series B:Biological Sciences,2006,361(1470):1007-1022.
[7] LUO Y H,SU Y,WRIGHT A D,et al.Lean breed Landrace pigs harbor fecal methanogens at higher diversity and density than obese breed Erhualian pigs[J].Archaea,2012,2012:605289.
[8] SAMUEL B S,GORDON J I.A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism[J].Proceedings of the National Academy of Sciences of the United States of America,2006,103(26):10011-10016.
[9] SAMUEL B S,HANSEN E E,MANCHESTER J K,et al.Genomic and metabolic adaptations ofMethanobrevibactersmithiito the human gut[J].Proceedings of the National Academy of Sciences of the United States of America,2007,104(25):10643-10648.
[10] BROCHIER-ARMANET C,BOUSSAU B,GRIBALDO S,et al.Mesophilic Crenarchaeota:proposal for a third archaeal phylum,the Thaumarchaeota[J].Nature Reviews Microbiology,2008,6(3):245-252.
[11] YOU J,DAS A,DOLAN E M,et al.Ammonia-oxidizing archaea involved in nitrogen removal[J].Water Research,2009,43(7):1801-1809.
[12] PESTER M,SCHLEPER C,WAGNER M.The Thaumarchaeota:an emerging view of their phylogeny and ecophysiology[J].Current Opinion in Microbiology,2011,14(3):300-306.
[13] SHI Y,HUANG Z,HAN S,et al.Phylogenetic diversity of Archaea in the intestinal tract of termites from different lineages[J].Journal of Basic Microbiology,2015,55(8):1021-1028.
[14] LI G N,XIA X J,TANG W C,et al.Intestinal microecology associated with fluoride resistance capability of the silkworm (BombyxmoriL.)[J].Applied Microbiology and Biotechnology,2016,100(15):6715-6724.
[15] LESZCZYSZYN J J,RADOMSKI M,LESZCZYSZYN A M.Intestinal microbiota transplant-current state of knowledge[J].Reumatologia,2016,54(1):24-28.
[16] BOJANOVA D P,BORDENSTEIN S R.Fecal transplants:what is being transferred?[J].PLoS Biology,2016,14(7):e1002503.
[17] TURNBAUGH P J,LEY R E,MAHOWALD M A,et al.An obesity-associated gut microbiome with increased capacity for energy harvest[J].Nature,2006,444(7122):1027-1031.
[18] KULECKA M,PAZIEWSKA A,ZEBER-LUBECKA N,et al.Prolonged transfer of feces from the lean mice modulates gut microbiota in obese mice[J].Nutrition & Metabolism,2016,13:57.
[19] DIAO H,YAN H L,XIAO Y,et al.Intestinal microbiota could transfer host Gut characteristics from pigs to mice[J].BMC Microbiology,2016,16:238.
[20] RAWLS J F,MAHOWALD M A,LEY R E,et al.Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection[J].Cell,2006,127(2):423-433.
[21] MCFALL-NGAI M.Love the one you’re with:vertebrate guts shape their microbiota[J].Cell,2006,127(2):247-249.
[22] WANG Z N,KLIPFELL E,BENNETT B J,et al.Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease[J].Nature,2011,472(7341):57-63.
[23] PANG X Y,HUA X G,YANG Q,et al.Inter-species transplantation of gut microbiota from human to pigs[J].The ISME Journal,2007,1(2):156-162.
[24] CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods,2010,7(5):335-336.
[25] BOKULICH N A,SUBRAMANIAN S,FAITH J J,et al.Quality-filtering vastly improves diversity estimates fromIlluminaampliconsequencing[J].Nature Methods,2013,10(1):57-59.
[26] EDGAR R C,HAAS B J,CLEMENTE J C,et al.UCHIME improves sensitivity and speed of chimera detection[J].Bioinformatics,2011,27(16):2194-2200.
[27] HAAS B J,GEVERS D,EARL A M,et al.Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J].Genome Research,2011,21(3):494-504.
[28] EDGAR R C.UPARSE:highly accurate OTU sequences from microbial amplicon reads[J].Nature Methods,2013,10(10):996-998.
[29] ZHANG H,DIBAISE J K,ZUCCOLO A,et al.Human gut microbiota in obesity and after gastric bypass[J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(7):2365-2370.
[30] GACI N,BORREL G,TOTTEY W,et al.Archaea and the human gut:new beginning of an old story[J].World Journal of Gastroenterology,2014,20(43):16062-16078.
*Corresponding author, senior livestock engineer, E-mail: yanghua806@hotmail.com
(责任编辑 王智航)
Intestinal Archaea Community Structure Analysis of Mice Transplanted withJinhuaand Landrace Pig Feces
XIAO Yingping1WANG Junjun2,3LI Tiantian2,3TANG Biao1HE Xiangxiang1LI Kaifeng1YANG Hua1*
(1.InstituteofQualityandStandardforAgro-Products,ZhejiangAcademyofAgriculturalSciences,Hangzhou310021,China; 2.BeijingAdvancedInnovationCenterforFoodNutritionandHumanHealth,Beijing100193,China; 3.CollegeofAnimalScienceandTechnology,ChinaAgriculturalUniversity,Beijing100193,China)
This experiment was conducted to explore the differences of the intestinal archaea community structures between the obese breedJinhuapig and the lean breed Landrace pig, and to investigate the changes of the archaea community structure during the inter-species fecal microbiota transplantation (FMT). The fresh feces ofJinhuaand Landrace pigs were collected and introduced into broad spectrum antibiotics treated germ-free mice guts by gavage, respectively. The total genomic DNA was extracted from the pig fecal samples and the transplanted mice cecal contents, and was used as templates to amplify the V4 region of the 16S rRNA gene by PCR with the universal primers of archaea. The PCR products were then subjected to high-throughput sequencing on an Illumina HiSeq sequencing platform. The obtained sequences were subsequently processed and analyzed with a series of software such as QIIME. The results showed as follows: at the phylum level, three main phyla of archaea, Thaumarchaeota, Euryarchaeota, and Crenarchaeota, were found in pig fecal samples and transplanted mice cecal contents, with Thaumarchaeota as the dominant phylum; while at the genus level, five known genera of archaea were observed, withCenarchaeumas the dominant genus. The abundance of phylum Euryarchaeota inJinhuapig’s feces was higher than that in Landrace pig’s feces. Thaumarchaeota dominate the archaea community structure of transplanted mice cecal contents, with abundance of more than 70%, and Thaumarchaeota mainly consisted ofCenarchaeum. It was observed that no significant difference was exhibited between the two groups of mice at the phylum and genus levels (P>0.05), but the operational taxonomic unit (OTU) based principal coordinate analysis showed a good aggregation within each group. It is concluded that genusCenarchaeumof phylum Thaumarchaeota is the dominant archaea in feces ofJinhuaand Landrace pigs and cecal contents of mice recipients. Phylum Euryarchaeota is more abundant inJinhuapig’s feces than in Landrace pig’s feces, and there is difference in archaea community structures between their respective mice recipients at the OTU level.[ChineseJournalofAnimalNutrition, 2017, 29(6):1895-1903]
archaea;Jinhuapig; Landrace pig; fecal microbiota transplantation; high-throughput sequencing
10.3969/j.issn.1006-267x.2017.06.010
2016-12-01
浙江省自然科学基金重点项目(LZ15C170001);北京食品营养与人类健康高精尖创新中心开放基金;国家自然科学基金(31402083)
肖英平(1984—),男,江西兴国人,助理研究员,博士,研究方向为畜禽肠道微生物。E-mail: ypxiaozju@126.com
*通信作者:杨 华,高级畜牧师,硕士生导师,E-mail: yanghua806@hotmail.com
S828
A
1006-267X(2017)06-1895-09
猜你喜欢
土壤学报(2022年3期)2022-08-26
河南畜牧兽医(2022年11期)2022-07-13
大自然探索(2022年5期)2022-07-11
今日农业(2022年7期)2022-07-06
知识就是力量(2022年6期)2022-06-16
当代畜禽养殖业(2022年1期)2022-03-29
现代畜牧科技(2021年5期)2021-07-20
江苏农业科学(2019年11期)2019-07-22
湖南畜牧兽医(2016年1期)2016-06-05
扬子江(2016年1期)2016-05-19
