
家族性噬血细胞性淋巴组织细胞增生症2例并文献复习
2017-06-22 14:23罗荣华王绪栋王兆辉王小丽
山东第一医科大学(山东省医学科学院)学报 2017年6期
孙 燕 罗荣华 冯 利 王绪栋 王兆辉 王小丽
(泰山医学院附属泰山医院儿科,山东 泰安 271000)
家族性噬血细胞性淋巴组织细胞增生症2例并文献复习
孙 燕 罗荣华 冯 利 王绪栋 王兆辉 王小丽
(泰山医学院附属泰山医院儿科,山东 泰安 271000)
目的 报道2例家族性噬血细胞性淋巴组织细胞增生症(FHL)患儿,并复习相关文献。 方法 回顾性收集、分析我院收治的2例FHL患儿的临床资料。 结果 例1患儿与其父有相同的UNC13-D基因杂合突变,其兄于1岁2月死于噬血细胞性淋巴组织细胞增生症(HLH),当时未行相关基因检测,复习相关文献推测其兄应亦患FHL;例2患儿与其父有相同的PRF1基因杂合突变位点,其母亲存在PRF1不同基因位点的杂合突变;例1患儿因家长放弃治疗离院,随访已死亡,例2患儿行HLH-2004方案化疗,化疗40周后获得完全缓解停药,但停药2月后复发,目前仍按HLH-2004方案化疗中。结论 例1患儿家庭中有3例FHL,有患儿与其兄2例发病,其父目前未发病;例2患儿家庭中亦有3例可诊断为FHL,患儿发病,其父母目前均未发病;FHL预后差;儿童HLH应及时行HLH相关的基因检测明确诊断并积极治疗;成人FHL未发病者应追踪随访。
噬血细胞性组织细胞增生症;家族性;治疗;基因检测
噬血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis, HLH)又称噬血细胞综合征(hemophagocytic syndrome, HPS),是一组由活化的淋巴细胞和组织细胞过度增生但免疫无效,引起多脏器高炎症反应的综合征;根据国际组织细胞协会诊疗指南,HLH分为原发性和继发性两种类型[1],原发性噬血细胞综合征包括家族性噬血细胞综合征(FHL)和免疫缺陷相关性噬血细胞综合征(iHLH)。该病起病急、病情进展迅速、死亡率高。现报道我院确诊的2例FHL患儿,并复习相关文献。
1 诊断标准[2]
详见中华医学会儿科学分会血液学组:噬血细胞性淋巴组织细胞增生症诊疗建议。
2 临床资料
例1患儿:男,6个月,汉族,因发热、咳嗽1周余入院。查体:神志清,精神欠佳,前囟平,咽充血,双肺呼吸音粗,可闻及干湿性啰音,心律齐,心音有力,未及杂音,腹软,肝肋下8 cm,质韧,脾肋下6 cm,质韧,神经系统无明显阳性体征。实验室检查详见表1;进行遗传代谢性疾病筛查,未发现与临床表型相关的变异;完善HLH基因检测提示患儿与其父有相同的UNC13-D基因杂合突变(详见表2)。因家长放弃治疗离院,随访已死亡。
既往史:该患儿出生时曾患“新生儿败血症,新生儿ABO溶血病,巨大儿”在当地住院治疗9天出院。家族史:患儿之兄在14月龄余死于HLH,当时未行相关基因检测,复习相关文献,推测亦患FHL;患儿之父:男,30岁,目前无任何HLH相关临床表现。
例2患儿:男,1岁4月,汉族,因反复发热近1月余入院,查体:神志清,精神欠佳,咽充血,双肺呼吸音,未闻及干湿性啰音,心律齐,心音有力,未及杂音,腹软,肝肋下6 cm,质韧,脾肋下4 cm,质韧,神经系统无明显阳性体征。实验室检查详见表1;进行遗传代谢性疾病筛查,未发现与临床表型相关的变异;完善HLH基因检测提示患儿与其父有相同的PRF1基因杂合突变位点,其母亲存在PRF1不同基因位点的杂合突变(详见表3)。入院后行HLH-2004方案[2]化疗(地塞米松、依托泊苷、环孢素),治疗40周后获得完全缓解后停药,但停药2月后复发,目前仍按HLH-2004方案化疗中。
既往史及家族史:无特殊;患儿之父:32岁,目前无任何HLH相关临床表现;患儿之母:29岁,目前无任何HLH相关临床表现。

表1 两例FHL患儿的实验室检查结果
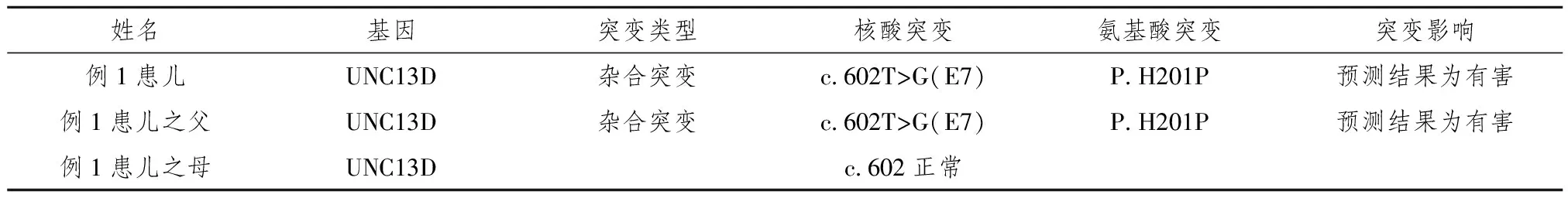
表2 例1患儿及父母HLH基因结果(北京德易东方转化医学研究中心提供)
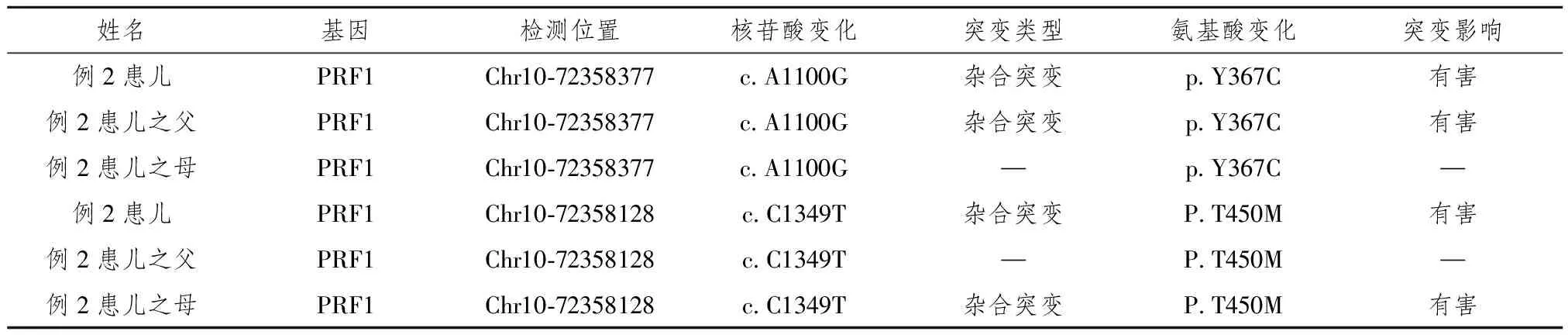
表3 例2患儿与父母HLH基因分析(北京迈基诺基因科技有限责任公司提供)
3 讨 论
HLH是一种以全身性组织细胞活化为特征的临床综合征.以淋巴细胞与巨噬细胞增殖活化为特点,并释放大量细胞因子,造成机体脏器功能损伤。Risdal等于1979年首次报道,HLH可于任何年龄人群发病,每年发生率约1/5000,其起病急骤、死亡率高,且易误诊[3-4]。FHL是常染色体或X连锁隐性遗传性疾病,多见于婴幼儿,90%患儿发病年龄<2 岁,死亡率较高,预后较差[5],发病率约为0.12/100000[6], 该病的诊断需要阳性家族史或做基因分析检测,因此临床上诊断FHL较困难。
目前已相继证实5种FHL相关的突变基因[7],部分患儿无家族史。根据发病机制,FHL目前已确定5型。FHL-1 的突变位点为9q21.3~22,但相关基因及功能蛋白表达尚未明确[8]。FHL-2 发病原因为穿孔素基因 (PRF1) 突变,定位于染色体 10q21~22[9],FHL中约30%由PRF1基因突变所致[10],本研究例2患儿即为PRF1基因突变所致,在国内亦有相关该型报道[11-12]。迄今已发现PRF1有70多个不同突变类型,RPF1突变使穿孔素蛋白表达缺失或减少,穿孔素功能不全,使NK细胞和细胞毒功能受损[13],部分突变可影响PRF1对靶细胞膜穿透能力[14]。FHL-3 的突变基因为UNC13-D[15],定位于染色体17q25,其基因产物 Munc13-4 蛋白,在细胞毒性颗粒的出胞中发挥重要作用。本研究例1患儿属于该类型。FHL-4 相关突变基因为突触融合蛋白11(syntaxin 11,STX11),定位于 6q24,编码产物为突触融合蛋白,该蛋白在细胞内转运中起重要作用。FHL-5是X连锁的隐性遗传,由编码Munc18-2的突触融合蛋白绑定蛋白2(syntaxin-binding-protein 2,STXBP2)基因编码 ,其位点为 19p13,在溶解性颗粒胞吐过程中发挥关键作用[15-16],在国内有相关该型报道[11]。
依据2002年修订的HLH的诊断标准[2],例1患儿及其父、例2患儿及父母均可明确诊断为FHL,复习相关文献推测例1患儿其兄应亦患FHL。根据相关的基因分析,例1患儿诊断为FHL-3型,该家庭中2例发病(该患儿及其兄),1例(患儿之父)未发病。 FHL-3型于2003年由Feldmann等[17]首先发现,源于编码Munc13-4蛋白的UNC13-D基因突变,定位于染色体17q25。UNC13-D突变占FHL的30%~35%[18]。 UNC13-D基因编码蛋白Munc13-4。Munc13-4蛋白主要参与囊泡引导及胞吐过程,是参与囊泡启动蛋白家族中的一员;UNC13-D突变损伤了囊泡启动及溶细胞酶释放,使细胞毒性颗粒吐胞功能障碍;Munc13-4蛋白不仅有助于细胞毒性T细胞内体微泡形成,调节细胞毒性颗粒分泌,还可能起调节血小板或多核白细胞颗粒分泌作用。例2患儿可诊断为FHL-2型,例2患儿有2个突变位点分别来自于患儿父亲及母亲,例2患儿及父的PRF1杂合突变位点已有文献报道,但患儿之母PRF1杂合突变位点尚未报道,可能为一新型的PRF1基因杂合位点。穿孔素通过在靶细胞上形成跨膜通道,使颗粒酶等进入靶细胞引起靶细胞的凋亡。PRF1突变使穿孔素的合成数量明显减少或合成的突变蛋白功能发生变化或两者皆有。HLH是一种异质性疾病, FHL患者不一定在早期发病,甚至有报道FHL发生在62岁的成年人[19-20],因此对于例1患儿父亲、例2患儿父母均需密切随访,做到早发现、早诊断、早诊断,降低死亡率。
HLH的治疗主要包括支持治疗、化学治疗及造血干细胞移植等,国内报道的采用HLH-1994或2004方案治疗的有效率在31.7%~56.1%[21-24],FHL的预后差,唯一根治方法是化疗缓解后予以造血干细胞移植,例1患儿家长放弃治疗后死亡,例2患儿经HLH-2004方案治疗后获得完全缓解,但停药2月后复发,均印证了这一点。FHL患儿诊断后如不治疗,均在短期内死亡,儿童HLH患儿应及时行HLH相关基因检测明确诊断,并早期干预,提高生活质量,降低死亡率。但目前仍有20%~50%的原发性HLH病例无明确基因异常[25]。故深入研究 FHL疾病相关的基因突变,建立具有确定诊断意义的基因诊断方法是今后努力的方向。
[1] Henter J I, Horne A, Arico M, et al.HLH-2004:Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J]. Pediatr Blood Cancer,2007,48:124-131.
[2] 中华医学会儿科学分会血液学组,噬血细胞性淋巴组织细胞增生症诊疗建议[J];中华儿科杂志,2012,,50(11):821-825.
[3] Janka GE.Hemophagocytic syndromes[J].Blood Rev,2007,21(5):245-253.
[4] Domachowske J B. Infections triggers of hemophagocytic syndrome in children.[J]. Pediatric infect Dis J,2006,25:1067-1068.
[5] Janka GE.Familial and acquired hemophagocytic lymphohistiocytosis[J]. Eur J Pedictr,2007,166,(2):95-109.
[6] Janka G, Zur Stadt U.Familial and acquired hemophagpcytic lymphohistiocytosis[J]. Hematol Educ Program, 2005, 2005: 82-88.
[7] Pachlopnik S J,Cote M,Menager M M,et al. Inherited defects in lymphocyte cytotoxic activity.[J]. Immunol Rev,2010,235:10-23.
[8] Ohadi M,Lalloz MR,Sham P,et al.Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping[J].Am J Hum Genet,1999,64 (1) : 165-171.
[9] Stepp S E,Dufourcq-Lagelouse R,Le Deist F,et al.Perforin gene defects in familial hemophagocytic lymphohistiocytosis[J].Science,1999,286( 5446) : 1957-1959.
[10] Grom AA. Macrophage activation syndrome and reactive hemophagocytic lymphohistiocytosis the same entities[J].Curr Opin Rheumatol, 2003, 15( 5 ):587-590.
[11] 王芳,何学鹏,陈惠仁.2 例罕见家族性噬血细胞综合征病因分析[J].临床血液学杂,2013,16(11):766-767.
[12] 冯春月,王秀敏,董关萍,等.家族性噬血细胞综合征1例报告[J].临床儿科杂志,2011,29(7): 690-691.
[13] Pachlopnik S J,Cote M,Menager MM,et al.Inherited defects in lymphocyte cytotoxic activity[J]. Immunol Rev,2010,235(1):10-23.
[14] Voskoboinik I,Thia MC,De Bono A,et al. The functional basis for hemophagocytic lymphohistiocytosis in a patient with coinherited missense mutations in the perforin PFN1 gene [J].Exp Med ,2004,200(6):811-816.
[15] De Saint Basile G,Menasche G,Latour S.Inherited defects causing hemophagocytic lymphohistiocytic syndrome[J].Ann N Y Acad Sci,2011,1246: 64-76.
[16] Zur Stadt U,Beutel K,Kolberg S,et al.Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: Molecular and functional analyses of prf1,unc13d,stx11,and rab27a[J].Hum Mutat,2006,27( 1) : 62-68.
[17] Feldmann J, Callebaut I,Raposo G,et al.Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis FHL3[J].Cell,2003,115(4):461-473.
[18] Pachlopnik SJ,Cote M,Menager MM,et al. Inherited defects in lymphocyte cytotoxic activity[J] .Immunol Rev ,2010,235 (1):10-23.
[19] Clementi R,Emmi L,Maccario R,et al.Adult onset and atypicl presentation of hemophagocytic lymphohistiocytosis in siblngs carrying PRF1 mutations[J]. Blood, 2002,100:2266-2267.
[20] Nagafuji K,Nonami A,Kumano T, et al.Perforin gene mutations in adult-onset hemophagocytic lymphocytosis[J].Haematologica,2007,92:978-981.
[21] 肖莉,宪莹,戴碧涛,等,HLH-2004方案治疗83例EB病毒相关噬血淋巴组织细胞增生症患儿疗效分析[J].中华血液学杂志,2011,32:668-672.
[22] 郭霞,李强,周晨燕,等.儿童噬血细胞综合征41例临床分析[J].中华血液学杂志,2007,28:449-453.
[23] Trottestam H, Horne A, Arico ,et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis;long-term results of the HLH-94 treatment protocol[J]. Blood,2011, 118:4577-4585.
[24] 广东省小儿噬血细胞综合征协作组.儿童噬血细胞综合征68例临床研究[J].中国小儿血液与肿瘤杂志,2009,14:20-22.
[25] Filipovich A, McClain K, Grom A.Histiocytic disorders: Recent insights into pathophysiology and practical guidelines[J]. Biol Blood Marrow Transplant, 2010,16: S82-89.
孙燕,女,主治医师,博士, 主要从事儿童血液/肿瘤工作 。
罗荣华,E-mail:rh6312@163.com.女,主任医师。专业:儿童血液及肿瘤。
R551.3
B
1004-7115(2017)06-0686-03
10.3969/j.issn.1004-7115.2017.06.036
2017-03-19)
猜你喜欢
种子(2021年3期)2021-04-12
中国现代医药杂志(2020年10期)2020-12-14
国际放射医学核医学杂志(2020年2期)2020-05-30
中成药(2017年5期)2017-06-13
哈尔滨医药(2016年1期)2017-01-15
外语教学理论与实践(2016年1期)2016-06-11
中学生理科应试(2016年7期)2016-05-14
现代检验医学杂志(2015年6期)2015-02-06
中国药业(2014年4期)2014-05-09
华东理工大学学报(自然科学版)(2014年1期)2014-02-27
