
小RNA高通量测序数据分析方法
2017-06-05 15:00李佛生王胜华
生命科学仪器 2017年2期
彭 骅, 李佛生,王胜华
小RNA高通量测序数据分析方法
彭 骅1,2, 李佛生1,王胜华1
(1.四川大学生命科学学院,成都,610064;2.中国科学院遗传与发育生物学研究所,北京 100101)
本文应用Perl语言和MySQL数据库构建了小RNA高通量测序数据分析平台,以4个水稻数据集为分析对象,详细介绍了小RNA高通量测序数据的处理方法和流程。我们以MSU 6.1水稻基因组为参考,构建了该版本的全基因组结构及已知ncRNAs位点信息数据库,结合Perl脚本可以实现小RNA在基因组上的详细定位与统计,同时我们从数据库中提取已知pre-miRNAs表达特征,设计了一个新的miRNAs挖掘方法,该方法可以筛选出大量的新miRNAs,其中已知miRNAs命中率可以达到98%。针对水稻小RNA种类的多样性,我们对miRNAs和endo-siRNAs的鉴别也给予了探讨和说明。本文设计的高通量测序数据分析平台,方法简单高效,以数据库作为存储和查询媒介,能够实现多位点reads的分析,可以得到灵活多样的数据统计结果。依照本文的方法同样可以构建其他模式物种的小RNA数据分析平台,在高通量测序逐渐普及的将来,本文的方法对中小实验室建立自己的数据分析平台具有实践指导意义。
小RNA(small RNAs);非编码RNA(ncRNAs);微小RNA(microRNAs,miRNAs) 内源小干扰RNA(endo-siRNAs);小RNA高通量测序(small RNA-Seq)
引言
小RNA(small RNAs)主要指长度在18-30nt的一类非编码RNA(ncRNAs),在真核生物中,具有基因表达调控功能的小RNA主要有微小RNA(microRNAs,miRNAs)﹑内源小干扰RNA(endo-siRNAs)和piwi干扰RNA(piRNAs).piRNA长度集中在26-31nt,目前只在动物的生殖系细胞及干细胞中被发现,其主要功能是参与转座子的沉默[1,2]。
miRNAs和endo-siRNAs长度主要集中在20-24nt.miRNAs在动植物和微生物中都普遍存在,目前在miRBase 14数据库中已包含115个物种的12627条记录[3]。 在细胞质中,miRNAs与AGO1等蛋白形成RISC 复合体(RNA-induced silencing complex), RISC通过miRNA与特定的mRNA靶基因互补配对,在配对区域的中间位置,AGO1通过对mRNA的切割促使其降解或者通过翻译抑制实现转录后调控[4-6]。 据估计一个物种中约1/3的基因会受到miRNA的调控[7],大量的实验也表明miRNAs参与了诸多生命过程的调控,例如细胞周期,细胞分化,组织器官的发生,营养代谢,信号途径以及对外界生物的非生物的环境的反应[9-12];同时,miRNAs在生产实践与临床治疗上也具有很大的应用前景[13,14]。 小分子干扰RNA(siRNAs)最初在植物转录后基因沉默现象中被报道,长度在20-25nt,来自外源的双链核糖核酸(dsRNA)切割产生[15]。随着小RNA研究的深入发展,大量的endo-siRNAs被发现,在植物体内,endo-siRNAs目前可以分为三种类型:(1)transacting siRNAs(ta-siRNAs),21nt,功能与miRNAs类似,与AGO1或AGO7组成RISC 复合体参与转录后调控。 (2) Naturalantisense siRNAs(nat-siRNAs),21nt和24nt, 21nt的nat-siRNAs功能与miRNAs类似,参与转录后调控.(3)Repeat-associated siRNAs(ra-siRNAs),24nt,参与染色体水平的基因沉默﹑抑制转座子的转座[16-18]。 显然, 生命体内还存在许多具有重要功能的small RNAs.如何鉴定与发现这些RNA是值得人们思索的重要问题。
以往用于寻找miRNAs等小RNA的方法有实验克隆法,计算机预测法[19-23]。 克隆法可以直接用于鉴定新小RNA,是初期发掘小RNA的常用方法,不足之处是实验周期较长,对低表达的小RNA的发现能力十分有限。 计算机预测法多是针对某一已知的小RNA特征设计算法,从全基因组或EST数据库中快速发掘大量潜在的小RNA,一定程度上弥补了克隆法的缺点,然而,预测的小RNA最终还需要实验证明,同时计算机预测法对新类型小RNA的发掘能力十分有限。 随着第二代高通量测序技术的问世,以测序为中心的功能基因组学研究开始全面展开[24,25],其中的小RNA高通量测序(small RNA-Seq)技术开始逐渐取代原始的小RNA发掘法方法,该法具有速度快﹑成本低﹑覆盖度深等多方面的优点,对鉴定与发现生命体内的小分子RNA及其功能与机理研究起极大的推动作用[26-28]。从最初应用454技术建立的谷类小RNA数据库(CSRDB,Cereal small RNAs Database)[29],到应用Illumina/Solexs技术建立的拟南芥小RNA计划[30](ASRP,Arabidopsis Small RNA Project),small RNA-Seq已被广泛应用于特定发育阶段全基因组水平上的小RNA的鉴定与发现[31-34]。 然而,在真核生物内小RNA种类繁多,高通量测序产生的数据量巨大,例如,一个水稻小RNA样本的Illumina测序数据可达2G大小,含有约1200万个测序片段(reads)读数,如何有效快速的处理这些数据,深入挖掘未知小RNA,是小RNA高通量测序数据分析的一个主要问题[35]。
目前小RNA高通量测序数据的挖掘能力还十分有限,已发表文献中所挖掘出的小RNA也只是数据中很小的一部分,而用于数据挖掘的综合软件也不多见,多是针对某些特殊需求设计的单一功能软件。 目前的开源软件主要有LeARN[36]﹑miRDeep[37]﹑CASHX[34]等,LeARN是基于perl语言的已知ncRNAs注释程序,miRDeep是基于perl语言的miRNAs挖掘程序,CASHX是拟南芥小RNA计划组开发的,是基于perl和MySQl的存储程序,它的功能和数据库结构比较简单。 而目前少数几种可用于高通量数据分析的商业软件价格相对昂贵,中小实验室难以负担,并且单纯依赖商业软件的分析结果往往又不能满足客户的各种要求。本文从实际应用出发,为科研人员详细介绍了高通量数据的分析方法。 我们采用Perl语言和MySQL数据库构建了小RNA高通量测序数据分析平台。 该平台设计简单科学,效率和功能强大,能够被大多数的中小实验所采用;可以实现多位点reads分析,而这部分数据在以往的分析中常常是被过滤掉的;可以实现数据的综合分析,得到灵活多样的统计结果;同时数据库本身具有很强的扩展能力,可以为各种小RNA及未知小RNA的挖掘提供支持,利用该平台我们也成功地挖掘出了一批新miRNAs。
2 材料和方法
2.1 实验平台及软件
本实验平台配置:IBM P615小型服务器,2核Power 1.45G处理器,8G内存,fedora 10 ppc 64位操作系统,MySQL 5.1数据库,用于数据库存储与查询;Hp dx2390 台式机,2核inter 2.8G处理器,4G内存,fedora 10 x86 64位操作系统,Perl 1.6,主要用于数据处理。
使用的软件有:Perl脚本,用于多个步骤中的数据处理;Perl-DBI模块主要用于MySQL数据表之间查询与更新;phpMyAdmin用于MySQL数据库管理和可视化查询;Soap[38]软件用于映射tags到参考基因组;RFAM/infernal用于已知ncRNA收集;dChip[39]用于芯片数据分析;RNAhybrid[40]用于miRNAs靶基因筛选;RNALfold[41]及miPred[41] 用于miRNAs二级结构分析。
2.2 小RNA高通量数据
2.2.1 实验数据集
本实验使用的数据集(表1)均为Illumina平台的水稻小RNA测序数据,sample1-3来源于NCBI的GEO数据库,sample4个来源我们实验室的测序结果,数据集在选择上并没有实验设计上的考虑,原因是能够获得到的原始数据集的数量非常有限,截止到目前,GEO数据库中仅有13个水稻高通量测序数据集。

Table1 Small RNA-Seq Datasets
2.2.2 原始数据格式
实验样品涉及的三种原始数据格式(表1),其均可以由Illumina测序平台生成,不同之处如下:
Scarf格式(图1a):每个read用单行表示,序列前面的部分表示实验编号和read所处的阵列位置;序列5’→3’排列,测序结果直接对应待测定的样本RNA或DNA序列,低质量的核苷酸用N表示;序列后边数字(-5~40)表示对应的序列质量,数值越小表示质量越不可靠,由于SBS测序法技术上的限制,随着测序长度的增加测序结果就越不可靠,所以Illumina平台的read长度一般控制在40nt以下。 Scarf格式是Fastq的压缩格式,数据较小便于传输,序列质量便与观察。

Fig.1 small RNA-Seq raw data format
Fastq格式:每个read都用四行表示(图1b),第1行和第3行为序列标识,含义同上;第2行为reads序列;第四行表示序列质量,Sanger-fastq序列质量采用33位至126位的ASCII 码编码,Illumina-fastq序列质量采用59位至126位的ASCII 码编码。
2.2.3 原始数据特点
(1)在一个实验的数据集中所有reads长度均相同,长度一般在40nt以下。
(2)Reads 3’端包含接头序列片段,长度不定。 例如,实验样本分离18-28nt范围的小RNA,测序长度为33nt,则每个read的3’端有5-15nt长度不等的接头序列。
(3)相同read 的数量可以直接代表该序列的表达量。
(4)reads数据量大,一般一个small RNA-Seq数据集包含的reads数不低于500万个。
2.2.4 高通量数据处理中的相关概念
由于reads的数量巨大,去除接头后的,我们可以把序列相同的reads合并,称为一个序列标签(tags),tags映射(mapped)到基因组后,位点相互重叠的tags我们可以聚集成簇(cluster),在一定距离内相互邻近的cluster我们可以进一步归为一个假设转录单元(unit)(图3)。
Read映射到基因组的位点我们称为hits,大多数reads 的位点是唯一的(即hits=1),这些reads在后续分析中比较容易处理,而hits>1的reads在以往的分析中往往被过滤掉,实际上很多miRNAs在基因组中是多位点的,其它类型ncRNAs中多位点现象更是普遍,目前多位点reads的定位和处理也是高通量测序中急需解决的一个基本问题,本文根据表达有效性对多位点reads进行过滤,根据上下游表达环境进行位点定位,可以实现大部分多位点reads的处理。
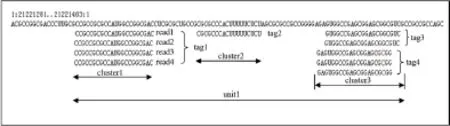
Fig.2 Illustration: read, tag, unit
2.3 参考基因组数据库
水稻有两个亚种:Japonica 和Indica,目前的基因组注释版本有Japonica:IRGSP Build5(2008-6-26更新),MSU v6.1(2009-6-3);Indica:BGI-9311(2006-12-28)。 由于MSU v6.1注释较为完整,在本文作为参考基因组[43]。 MSU v6.1共有56797个基因位点,其中32%的基因位点有GO注释,28.6%的基因位点与转座元件(TEs)相关。 我们根据MSU v6.1的基因组注释信息,从新划分了基因组结构(图3),构建了用于高通量数据分析的Rice数据库(图4)。
2.3.1 基因组结构信息表的构建
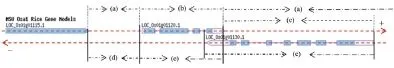
Fig.3 Illustration:genomic structure(a)intergenics on ssDNA.(b) genes on ssDNA.(c)gene_antisense on ssDNA.(d)intergenics on dsDNA.(e) genes on dsDNA
如果将DNA单链分别对待,例如以“+”链为例,可将单链分为基因间隔区(图3a)和基因编码区(图3b),在某些基因间隔区中还包含“-”链基因的反义链区(图3c);如果把DNA双链为整体对待,可将DNA分为基因位点间隔区(图3d)和基因位点区(图3e).
在MSU v6.1的注释文件中,我们根据已知的(b)﹑(d)位点信息构建了(a)的位点信息,分别导入到本地rice数据库的genes表﹑intergenic表和fb_intergenic中表。由于某些基因存在重叠情况(图3),我们创建了重叠基因信息表genes_cross表。对于基因内部的结构信息(内含子﹑外显子﹑3’﹑5’端),将其存储在feature表中。其中genes表和fb_intergenic表(表2)是基因组数据库的核心表,与核心表对应的map_genes表和map_ fbintergenic表用于存储不同样本中tags所映射的genes及fb_ intergenic的表达情况。

Fig.4 Rice genome database structure

Table2 fb_intergenic table structure
2.3.2 已知miRNAs、ncRNAs和marker信息表的构建
如果基因组的注释比较全面,则有关的注释信息可以直接从注释文件中获取,如果注释不够全面或者达不到实验目的要求,则需要自己收集相应的序列信息再通过blastn映射到基因组,本实验建立了6个相关注释信息表,如(图4)中所示,包括miR14表﹑ncRNA表﹑repeat表﹑FST表﹑MULE表﹑organelle表,其中将repeat﹑FST﹑MULE﹑organelle归类为基因标识(marker),三个表的信息来源及说明见(表3)。

Table3 miRNA、ncRNA and maker tables description
Repeat表存储水稻重复序列位点信息,来自MSU Oryza Repeat Database v3.1;FST表存储侧翼序列标签(FST,Flanking Sequence Tags),来自Tos17,T-DNA或Ac/Ds的插入突变位点侧翼序列;MULE表存储基因突变元件(MULE,Mutator like elements),来自突变基因或基因片段;organelle表存储细胞器插入片段(Organellar Insertions),来自水稻叶绿体和线粒体在基因组上的同源序列。我们选择这些序列作为marker不但可以对已知miRNAs的做起源分析,还可以用于未知的tags的深入分析。
与6个注释信息表相对应的6个map_*表(图4),用于存储不同样本在每个注释位点上表达差异信息。如map_miR14表中,每个已知的miRNAs分别存在4个tc字段﹑4个rc字段﹑4个h1rc字段,分别代表4个样本中的tags数量﹑reads数量﹑位点唯一的reads的数量。
2.4 样品tags数据库
本实验建立了四个样品数据库,每个数据库用于存储各自样品中的tags信息和tags在基因组上的映射信息,数据库结构如(图5)所示。结合(图3)所示的基因结构对tags映射的基因组类型进行分类并存储在不同的映射表中(表4).

Table4 Sample tables function description

Fig.5 Sample database structure
样品数据库的核心表为tags_map表﹑tags_to_cluster表和cluste_to_unit表。tags_map表中核心字段为r_count,hits,map_label,map_ncRNA,gff_label(表5),其字段说明如下。

Table5 tags_map table structure
tags_to_cluster表中核心字段为tc,rc,h1rc,map_label,ncRNA_lable,gff_label,all_tagsid_rc。其中tc存储cluster在一起的tags计数;rc是对所有reads计数;h1rc是对hits=1的reads计数;map_label,ncRNA_lable,gff_label来自tags_map的map_lalel,ncRNA_lable,gff_label的推导;all_tagsid_rc存储聚集在一起的tags的id等信息,我们根据tags聚集情况对多位点tags进行打分,评价其真实的转录位点。
除了四个样本数据库之外,本文还构建了一个HTS数据库,用于存储4个样品中reads数量大于3的tags记录,该数据库可以用于tags在不同样本中的表达差异分析。
2.5 高通量数据处理流程与方法
2.5.1 处理流程

Fig.6 Data flow(A) Basic processing.(B) New-miRNAs data mining
2.5.2 处理方法
1)数据基本处理.如(图6A)所示,包括步骤①-⑥,概述如下:
步骤①:reads接头去除。由于部分reads 3’端序列质量较低,错误的碱基较多,为去除接头带来一定难度,本实验采用的动态接头去除法,对末尾低质量的碱基进行递归以确保最大程度的去除接头。 与样品1﹑2的原作者处理结果相比较[32],我们的处理结果要优于前者。
步骤②:tags映射到基因组。 目前针对高通量测序开发的映射软件主要有soap(SOAPaligner)﹑ELAND﹑MAQ﹑Bowtie﹑GenomeMapper等,我们使用了华大基因组开发的soap 2。 0,其优点是可以输出多位点tags的所有映射位点信息,参数设置允许2个错配无Gaps。
步骤③:tags映射信息导入数据库。 由于多位点的tags的位点信息量过大,我们根据tags的表达的有效性进行筛选,hits 1-6的tags映射信息全部导入,hits 7-20的tags,将所含reads数量大于3的位点信息导入数据库,hits 21-50的tags,将所含reads数量大于10的位点信息导入数据库。 已知tRNA(Met_CAT)在基因组上的位点数最多(hits=50),所以我们将hits值大于50的tags不再导入数据库。 经过对多位点tags的筛选,映射到基因组的reads中约90%的数据被导入到数据库(表6)。
步骤④:tags位点结构归类。 将tags_map表中的位点信息与rice数据库中的genes﹑intergenic及fb_intergenic的位点信息进行匹配查询,匹配结果分别导入到不同的结构表中,同时更新tags_map表的map_label字段,该字段用不同的标识代表tags所在位点的基因结构属性。 我们按map_lable字段分组汇总即可获得tags在基因组上的分布情况。
步骤⑤:tags位点注释。 将各样本的tags_map中的位点信息与rice数据库的miR14和表ncRNA分别进行匹配查询,查询结果导入表map_miR14和表map_ncRNA,同时更新tags_map表的ncRNA_label字段;对4个marker表匹配查询结果分别导入map_repeat﹑map_FST﹑map_MULE﹑map_organelle 4个表,同时将各类ncRNA的标识写入tags_map的gff_label字段。 我们对ncRNA_lable字段的汇总即可获得tags在已知ncRNA中的表达情况(图10),对gff_label字段的汇总即可得到tags在已知marker上的分布情况。
步骤⑥: tags聚簇。 我们将表tags_map的位点信息聚集成簇,结果导入tags_to_cluster表;在将cluster信息聚集成单元,结果导入cluster_to_unit表。 生成cluster之后, 我们根据cluster包含的tags聚集特点对hits>1的tags进行定位处理。
2)新miRNAs挖掘。 如(图6B)所示,包括步骤(a)-(d),概述如下:
步骤(a):初步筛选。 通过对已知pre-miRNAs的表达特征的分析,设计算法,对tags_to_cluster表中没有映射到已知ncRNAs的cluster进行筛选,再从筛选的cluster中对tags进行筛选,筛选出来的tags称为假设的miRNAs(hyp-miRNAs)。
对cluster筛选的主要条件:18<cluster_length<50 AND (h1rc/tc>=5 OR rc/tc>=50);
对tags筛选的主要条件:18<tags_length<26 AND r_count/ hits>10 ;
步骤(b):靶基因过滤。 如对应的小RNA样本阶段存在相应的mRNA表达谱数据(如转录组芯片数据,数字标签测序数据),表达谱中下调的mRNA可以假设为miRNAs的靶基因(target),以这些靶基因为过滤材料,通过靶基因预测我们可以过滤出一大批符合miRNAs/target配对条件的hyp-miRNAs。
步骤(c):二级结构过滤。 提取hyp-miRNAs所在unit的RNA序列,如果unit长度小于80nt,则两边各取150nt。 我们采用RNALfold程序对提取的RNA进行了筛选,该程序可以在指定的长度(-L)内递归搜索二级结构最稳定的子序列,我们从最大长度开始以20nt为单位递减,但最短不小于80nt,该值作为 -L的参数,筛选满足无内环的发夹结构并含有tags位点的最长子序列,该子序列作为假设的miRNAs前体(hyp-premiRNAs)。
步骤(d):pre-miRNAs鉴定。 我们采用miPred程序对hyp-pre-miRNAs序列进行打分,一般得分在52以上的序列,miPred就认为其是真实的 pre-miRNAs,而真实的 pre-miRNAs上处于发夹结构茎区的表达量最高的tags我们就认为其是新的New-miRNA。
3 结果
按照图6所示的流程和方法,分别对四个样本数据集进行了处理,应用MYSQL查询语句可以从不同角度对每个数据表进行统计,这里主要从以下这几个方面展示处理结果。
3.1 基本数据处理结果
3.1.1 样本reads/tags数量统计
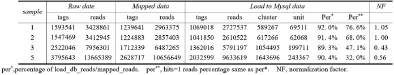
Table.6 sample reads / tags count
3.1.2 样本reads/tags长度分布
样品1,种子发育1-5天,分离18-25nt范围的小RNA。将去除接头后的数据和导入数据库的数据按reads/tags数量对长度做图(图7),图中raw_rc曲线比较规则,说明RNA样本完
由于处理过程多处优化,导入数据库中用于分析的数据比例比原始文献都要高[32]。如果进行样本间的表达差异比较,则reads计数需要做标准化处理,由于4个样本分离的小RNA长度有所不同,我们选取长度在17nt~28nt,tags包含的reads数大于3的所有reads计数做平均值标准化处理(表6)。整性较好;峰值出现在21﹑24,说明此长度的小RNA大量表达。
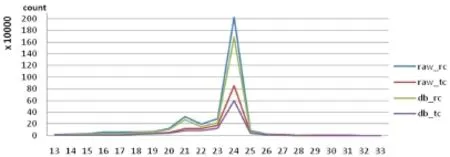
Fig.7 Sample1 reads/tags length distribution.raw_rc, reads count in raw data.db_tc, tags count of load mysql
样品4,花粉发育3核阶段,分离12-40nt范围的小RNA.将不同处理阶段的数据做图(图8),图中raw_rc曲 线相对不是很规则,说明样本完整性一般;将已知ncRNAs过滤后得到free_rc曲线。比较db_rc曲线,可以看出峰值16﹑19及30-39范围的reads基本消失,说明这些reads大多是已知的ncRNA降解片段;而free_rc曲线中仍然存在21-23nt及23-25nt两个峰,尤其24nt峰值很高,说明还有大量的未知small RNAs待挖掘。
3.1.3 Reads 位点分布图
根据样本数据库tagsid表中的hits字段分组,对r_count字段汇总,即可得到reads在基因组上的位点分布情况(图9),由图可知,不同组织的样本小RNA表达存在显着的差异.如样本1﹑2为同一组织,差异并不明显,而叶片与成熟花粉多位点小RNA比例很高,这说明该组织中大量位于repeat区域的小RNA表达,或者某些多位点小RNA大量表达,也可能为转录后扩增现象所致。
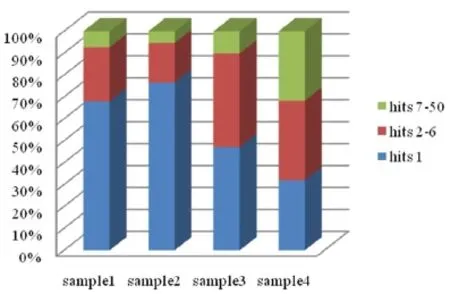
Fig.9 Sample1-4 reads hits distribution
3.1.4 Reads在基因组及已知ncRNA分布情况
根据tags_map表中的ncRNA_label字段分组,对r_count字段汇总,可以得到reads在已知ncRNAs中的表达情况(图10).由图可知,不同组织中的ncRNAs的表达存在显著差异,如叶片样本3中已知的miRNAs大量表达,占总reads的47.6%;在花粉样本4中,已知的tRNA大量表达,约占总reads的51.8%;图中intergenics﹑genes和gene_antisence部分是没有映射到已知ncRNAs的reads部分,其中intergenics和gene_antisence的比例可以代表样本中未知的有待挖掘的小RNA部分.
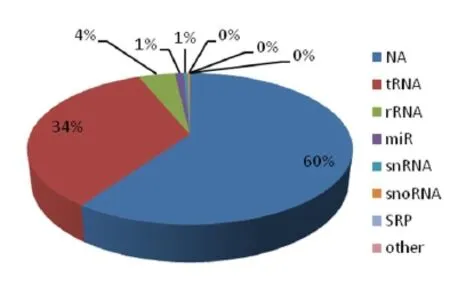
Fig.11 Sample4 reads distribution on ncRNAs NA, no mapped known ncRNAs reads
对样本4分析发现,大量的tRNA降级片段存在与30-39nt范围内(图8),因其他3个样本分离的小RNA长度都不超过30nt,所以我们对样本4中小于30nt的reads重新做了一个统计(图11),由图可见已知的tRNA仍然占了总量的34%,这说明花粉中tRNA确实存在大量表达,或者tRNA在样本4中发生了大量降解.
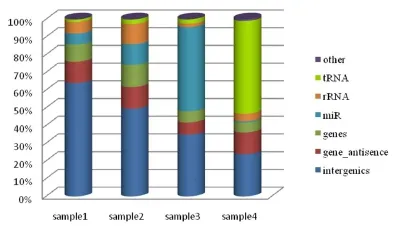
Fig.10 Sample1-4 reads distribution on ncRNAs and genome.miR,mapped pre-miRNAs or miRNAs.
3.2 已知miRNAs表达特征分析
3.2.1 已知ncRNAs的表达特征
如果一些tags片段是来自一个较长前体的随机降解,那么这些tags头尾重叠的概率较大,cluster处理后就很有可能聚集在一个cluster中,这样的一个cluster所包含的reads/tags值应该较小。 如果这些短的tags片段是来自前体的规则剪切,那么tags间重叠的概率就较小,剪切点两侧的序列在一个cluster中的概率就很小,这样的一个cluster的reads/tags值应该较大。基于这样的原理,对样品1-2的4类ncRNAs在tags_to_cluster表中的reads/tags比值进行统计,结果列于(表7)。
从结果中可以看出,pre-miRNAs的两类比值均较高(56.36,4.77),这说明pre-miRNA的剪切比较规则,这两类比值可作为pre-miRNAs前体的初级筛选条件。如将cluster长度限制在小于50nt,Pre-miRNA筛选数量可以占到总量的90%(307/342),这说明Pre-miRNA剪切所产生的不同位置的tags片段重叠程度很低,这也与miRDeep筛选方法中对PremiRNA剪切特点的假设相一致[37]。
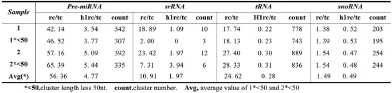
Table.7 the reads/tags ratio of ncRNAs in tags_to_cluster table
3.2.2 已知miRNAs信息统计
在miR14 表中有414个水稻已知miRNA前体(osa-premiRNAs)记录,451个水稻miRNA(osa-miRNAs)记录,从不同角度统计如下:(1)318个miRNAs归类为已知的56家族,133个miRNAs未归类为任何家族;(2)451个miRNAs共包含271个特异序列;(3)miRNAs长度分布:20nt-51个,21nt-340个,22nt-212个,24nt-12个;pre-miRNAs平均长度为140nt;(4)实验验证:未验证-145个,cloned-142个,Northern-87个,MPSS-23个,454-117个,Solexs-44个;(5)Pre-miRNAs在MSU v6.1 上共684个位点记录:181个位点位于repeat区域,11个位点位于FST区域,78个位点位于MULE区域。(6) miRNAs在MSU v6.1 上共731个位点记录:基因间隔区-521个,基因区-128个,基因反义链-87个。
3.2.3 已知osa-miRNAs表达图谱
将tags_map表ncRNA_label字段标识为“os01,os02”(表3)的记录按照长度分组对r_count字段进行汇总(hits>1的tags记录, r_count值取平均值),汇总结果经标准化处理,统计结果见(图12)所示。
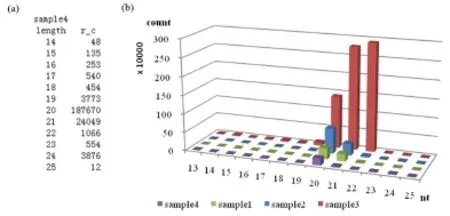
Fig.12 Sample1-4 osa-miRNAs reads-length distribution.(a)sample4 osa-miRNAs reads count group by length.(b)sample1-4 osa-miRNAs reads-length distribution

Table.8 the number of miRfam expression in diffident sample
这里我们将已知的56个osa-miRNAs家族和无家族归类的133个osa-miRNAs统称为miRfam,在map_miR14表中筛选reads计数大于3的miRfam数量,统计结果见(表8)所示。
我们按已知的56个osa-miRNAs家族(MIR)分组, 在map_ miR14表中对每个家族的miRNAs成员的表达量汇总后取平均值,在经过标准化处理作为MIR的表达量,最后筛选MIR表达量reads之和大于20的记录, 共计34个MIR,绘制表达差异图谱,见(图13)所示.
Fig.13 Sample1-4 osa-miRNAs family expression maps
3.3 新miRNAs挖掘结果
样本1﹑2中原作者共挖掘了39个新miRNAs(osa-miR1846至osa-miR1884)[32],在此之外,我们对原始数据从新进行了深入挖掘,如(图6B)所示。
步骤(a):经初步筛选共获取了1694个序列特异的hypmiRNAs和142个osa-miRNA。 已知在两个样本中表达量reads大于10的osa-miRNA共143个,初步筛选命中率为99%。
步骤(b):对应样本1﹑2(1-5DAF,6-10DAF),刚好收集到对应的0-1DAF﹑2-4DAF﹑5-10DAF和11-20DAF四个阶段的全基因组芯片数据[44],通过dchip分析获取了1089个表达值至少下调5倍的靶基因,采用RNAhybrid软件对hyp-miRNAs进行靶基因过滤,参数如下:
RNAhybrid -m 7000 -f 8,12 -u 1 -v 1 -e -32 -q hypmiRNAs -t down_target_genes > miR_target
参数-m 7000表示对靶基因搜索长度不超过7000nt;参数-f 8,12表示miRNA/target配对区域8-12的范围内为完全配对;参数-u 1表示允许一个内环;参数-v 1表示允许一个凸环;参数-e -32表示最小自由能小于-32。
对RNAhybrid输出数据我们做了进一步的筛选,保留配对区域上hyp-miRNAs 的5’突出不多于2nt,3’突出不多于4nt的配对结果,结果如下:1089个靶基因有985个存在匹配;1694个hyp-miRNAs保留了664个(其中149个位于genes位点,44个位于repeat位点,22个位于MULE位点),142个osa-miRNA保留了103个。
步骤(c):664个hyp-miRNAs提取到180个unit,RNALfold筛选后得到152个hyp-pre-miRNAs;103个osa-miRNA有102个满足RNALfold筛选条件。
步骤(d):miPred程序对hyp-pre-miRNAs进行打分验证,结果如下:152个hyp-pre-miRNAs中有135个可以通过miPred验证(图14);102个osa-miRNA全部可以通过验证;osamiRNA的平均得分值为74,135个新的pre-miRNAs平均得分值为73。
筛选过程中以已知的osa-miRNA为参照,步骤(a)﹑(c)对osa-miRNA的筛选命中率均可以达到99%,而步骤(b)的可变因素较多,很难估计筛选的准确性.如果我们跳过(b)选择(a)-(c)-(d)的处理流程,osa-miRNA的命中率可以达到98%.对样本3﹑4的挖掘结果我们将在其他文章中给予报道。
135个新pre-miRNAs,其中52个pre-miRNAs的miRNA和miRNA*中同时存在(图14a),其余的pre-miRNAs仅存在miRNA(图14b,我们选择表达量最高的tags作为miRNAs,青色标识)
详细的miRNA挖掘流程和打分标准,可以近一步参考作者已发论文[51]的方法部分。

Fig.14 New-pre-miRNA and miRNAs(a)new-pre-miRNA with aligned small RNAs tags(contain miRNA and miRNA*),the behind number are reads count, the predicted secondary structure shown in bracket notation.(b) new-pre-miRNA contain miRNA only.
4 讨论
Small RNA-Seq产生的数据量巨大,以往的一些方法中直接对reads进行分析[32,33],数据处理复杂度较高,处理结果以文本形式存储,不便于查询统计。 我们建立的方法主要由数据的基本处理与挖掘两部分组成。在基本数据处理中,按照“reads→tags→cluster→unit”的处理步骤逐步压缩数据的复杂度,并将各步骤的处理结果导入数据库,这样既可以实现数据间的关联,又便于查询统计。 在数据的分析中,我们建立了参考基因组数据库,分别从单链与双链的角度解剖分析基因组结构,这样可以得到tags映射位点的详细结构信息,便于数据的深入挖掘;同时,通过建立基因组marker数据表,很容易实现tags所处位点的注释和统计,并且数据库结构容易扩展,新的注释信息很容易导入数据库参与tags注释。 在以往分析中, hits>1的reads处理起来较复杂, 对这类数据的过滤会丢失许多有价值的信息, 我们通过对多位点数据的筛选﹑位点定位处理, 可以实现大部分有意义的多位点reads的分析, 很大程度上提高了数据的挖掘能力。
我们设计的新miRNAs挖掘方法,对已知的osa-miRNAs筛选命中率能够达到98%,可见该方法具有很高的有效性。该方法与目前的miRdeep方法相比较能够挖掘出更多的新miRNAs,这主要是由于miRdeep筛选方法过于严格所致[37]。一个真实的pre-miRNAs被DCL1剪切可以产生三类短片段,即miRNAs﹑miRNAs*和发卡结构环区片段,并且三类片段出现重叠的概率很低,miRdeep方法所挖掘的新pre-miRNAs中要求其所产生的miRNAs/miRNAs*同时在测序数据中存在(图14a),这种苛刻的过滤条件尽管确保了挖掘出的新的miRNA真实可靠,但同时也将不少有价值的信息给丢掉了。例如,我们就已知的pre-miRNAs作分析,发现只有50%的pre-miRNAs满足这个原则。我们采用靶基因筛选过滤的步骤,一方面确保筛选得到的miRNA存在具体的靶mRNA,另一方面拓宽了发现新miRNA的能力;因为有一些pre-miRNAs产生的miRNAs﹑miRNAs*和发卡结构环区片段的降解周期不一致,导致这三种片断不可能长时期同时存在。 除了通过mRNA表达谱获取潜在的靶基因,目前还可以通过直接测序获取,即降解组测序法,也称RNA末端平行分析(PARE,parallel analysis of RNA ends)[45-47],其基本原理是通过收集被AGO蛋白剪切的mRNAs片段,将这些片段进行扩增后测序,测序结果与假设的miRNAs进行匹配搜索,这样可以同时实现miRNAs和靶基因的验证。
通过对tags在基因结构及已知ncRNAs上的分布统计(图10),我们可以推断出有待挖掘的小RNA比例,我们发现未知小RNA中24nt所占的比例相当高(图8)。 已知在植物的DCL酶存在4种类型,DCL1可以产生20-24nt的miRNAs和21nt的natsiRNAs,DCL2产生24nt的nat-siRNAs,DCL3产生24nt的rasiRNAs,DCL4产生21nt的ta-siRNAs[17,18,48,49]。 对与外源病毒介导产生的dsRNA,DCL2﹑DCL3和DCL4均参与其剪切分别产生22﹑24 和21 nt的siRNAs[50]。 由此可见,同一长度小RNA的来源多样,尤其是24nt的小RNA,如何对24nt-RNA进行分辨也是数据分析中的一个难点。 已知三种类型endo-siRNAs前体都不具备显著的发夹结构,我们可以根据这个特点首先将大批的潜在miRNAs过滤出来;已知ta-siRNAs的前体较长,一个前体可以产生多个相邻的21nt的ta-siRNAs,根据这个特点,我们可以在culster_to_unit表中筛选较长的unit且tags长度以21nt为主,同时我们知道ta-siRNAs前体是某些miRNAs的靶基因,因此可以通过其前体与miRNAs的靶基因筛选进一步验证ta-siRNAs的真实性;已知ra-siRNAs的前体来源于repeat或TEs序列,我们可以在map_repeat数据库中所搜高表达repeat记录,并且其包含的tags长度以24nt为主;已知natsiRNAs的前体起源于基因重叠区,我们可以通过对映射到基因重叠区的tags进行筛选,并且tags长度以21nt和24nt为主。已知的endo-siRNAs前体dsRNA都会出现扩增现象,这样对cluster位点正负链上的表达情况分析可对endo-siRNAs做进一步验证。 本实验构建的数据库结构完全可以满足上述各类小RNA挖掘方法上的要求,我们构建了交叉基因的数据集,构建了包含转子序列的repeat数控库,在已知注释信息表的基础上,也很容易构建其相应反义链的信息表。进一步的实验中,我们将会对各类小RNA进行深入挖掘,同时我们也期待有新类型的小RNA能在数据发掘中被发现。
总之,我们设计的small RNA-Seq分析平台,具备综合的数据分析能力,能够得到灵活多样的统计结果,能够快速的对小RNA进行深入挖掘,数据库结构简单高效,可以被大多中小实验室所采用。
[1] Das P P, Bagijn M P, Goldstein L D, et al.Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline[J].Mol Cell, 2008.31(1): 79-90.
[2] Brennecke J, Malone C D, Aravin A A, et al.An epigenetic role for maternally inherited piRNAs in transposon silencing[J].Science, 2008.322(5906): 1387-1392.
[3] Jones S G, Saini H K, Dongen S V, et al.miRBase: tools for microRNA genomics[J].Nucleic Acids Res, 2008.36: D154-D158.[4] Williams A E.Functional aspects of animal microRNAs[J].Cell and Mol Life Sci, 2008.65(4): 545-562.
[5] Voinnet O.Origin, biogenesis, and activity of plant microRNAs[J].Cell, 2009.136(4): 669-687.
[6] Liu X H, Fortin K, Mourelatos Z.MicroRNAs: biogenesis and molecular functions[J].Brain Pathol, 2008.18(1): 113-121.
[7] Lewis B P, Burge C B, Bartel D P.Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets[J].Cell, 2005.120(1): 15-20.
[8] Poethig R S.Small RNAs and developmental timing in plants[J].Curr Opin Genet Dev, 2009.19(4): 374-378.
[9] Chen X M.Small RNAs and Their Roles in Plant Development[J].Annu Rev Cell Dev Bio, 2009.25: 21-44.
[10] Williams A E.Functional aspects of animal microRNAs[J].Cell and Mol Life Sci, 2008.65(4): 545-562.
[11] Rooij E V, Sutherland L B, Qi X X, et al.Control of stressdependent cardiac growth and gene expression by a microRNA[J].Science, 2007.316(5824): 575-579.
[12] Song L, Tuan R S.MicroRNAs and cell differentiation in mammalian development[J].Birth Defects Res C Embryo Today, 2006.78(2): 140-149.
[13] Ventura A, Jacks T.MicroRNAs and cancer: short RNAs go a long way[J].Cell, 2009.136(4): 586-591.
[14] Ochiya T.Micrornas as Therapeutic Targets: Potential Effect on Prostate Cancer Management[J].J Gene Med, 2009.11(12): 1143-1143.
[15] Hamilton A J, Baulcombe D C.A species of small antisense RNA in posttranscriptional gene silencing in plants[J].Science, 1999.286(5441): 950-952.
[16] Schwach F, Moxon S, Moulton V, et al.Deciphering the diversity of small RNAs in plants: the long and short of it[J].Brief Funct Genomics, 2009.8(6): 472-481.
[17] Vazquez F.Arabidopsis endogenous small RNAs: highways andbyways [J].Trends Plant Sci, 2006.11(9): 460-468.
[18] Brodersen P, Voinnet O.The diversity of RNA silencing pathways in plants[J].Trends Genet, 2006.22(5): 268-280.
[19] Yousef M, Showe L, Showe M.A study of microRNAs in silico and in vivo: bioinformatics approaches to microRNA discovery and target identification[J].Febs J, 2009.276(8): 2150-2156.
[20] Hou Y Y, Ying X M, Li W J.Computational approaches to microRNA discovery [J].Hereditas, 2008.30(6): 687-96.
[21] Berezikov E, Cuppen E, Plasterk R H.Approaches to microRNA discovery[J].Nat Genet, 2006.38 Suppl: S2-7.
[22] Huttenhofer A, Vogel J.Experimental approaches to identify non-coding RNAs[J].Nucleic Acids Res, 2006.34(2): 635-46.
[23] Kawaji H, Hayashizaki Y.Exploration of small RNAs[J].Plos Genet, 2008.4(1): e22.
[24] Wold B, Myers R M.Sequence census methods for functional genomics[J].Nat Methods, 2008.5(1): 19-21.
[25] Schuster S C.Next-generation sequencing transforms today's biology[J].Nat Methods, 2008.5(1): 16-18.
[26] Nobuta K, Venu R C, Lu C, et al.An expression atlas of rice mRNAs and small RNAs[J].Nat Biotechnol, 2007.25(4): 473-477.[27] Zhang X Y.The epigenetic landscape of plants[J].Science, 2008.320(5875): 489-492.
[28] Zhou X F, Sunkar R, Jin H, et al.Genome-wide identification and analysis of small RNAs originated from natural antisense transcripts in Oryza sativa[J].Genome Res, 2009.19(1): 70-78.
[29] Johnson C, Bowman L, Adai A T, et al.CSRDB: a small RNA integrated database and browser resource for cereals[J].Nucleic Acids Res, 2007.35: D829-D833.
[30] Backman T W, Sullivan C M, Cumbie J S, et al.Update of ASRP: the Arabidopsis Small RNA Project database.Nucleic Acids Res, 2008.36(Database issue): D982-985.
[31] Jones-Rhoades M W, Borevitz J O, Preuss D.Genomewide expression profiling of the Arabidopsis female gametophyte identifies families of small, secreted proteins[J].Plos Genet, 2007.3(10): 1848-1861.
[32] Zhu Q H, Spriggs A, Matthew L, et al.A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains[J].Genome Res, 2008.18(9): 1456-1465.
[33] Wang X, Elling A A, Li X Y, et al.Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize[J].Plant Cell, 2009.21(4): 1053-1069.
[34] Fahlgren N, Sullivan C M, Kasschau K D, et al.Computational and analytical framework for small RNA profiling by highthroughput sequencing[J].RNA, 2009.15(5): 992-1002.
[35] Pop M, Salzberg S L.Bioinformatics challenges of new sequencing technology[J].Trends Genet, 2008.24(3): 142-149.
[36] Noirot C, Gaspin C, Schiex T, et al.LeARN: a platform for detecting, clustering and annotating non-coding RNAs[J].Bmc Bioinformatics, 2008, 9: 21.
[37] Friedlander M R, Chen W, Adamidi C, et al.Discovering microRNAs from deep sequencing data using miRDeep[J].Nat Biotechnology, 2008.26(4): 407-415.
[38] Li R Q, Li Y R, Kristiansen K, et al.SOAP: short oligonucleotide alignment program[J].Bioinformatics, 2008.24(5): 713-714.
[39] Li C.Automating dChip: toward reproducible sharing of microarray data analysis[J].Bmc Bioinformatics, 2008, 9: 231.
[40] Kruger J, Rehmsmeier M.RNAhybrid: microRNA target prediction easy, fast and flexible[J].Nucleic Acids Res, 2006.34(Web Server issue): W451-454.
[41] Hofacker I L, Priwitzer B, Stadler P F.Prediction of locally stable RNA secondary structures for genome-wide surveys[J].Bioinformatics, 2004.20(2): 186-190.
[42] Jiang P, Wu H N, Wang W K, et al.MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features[J].Nucleic Acids Res, 2007.35(Web Server issue): W339-344.
[43] Yuan Q P, Ouyang S , Wang A H, et al.The institute for genomic research Osa1 rice genome annotation database[J].Plant Physiol, 2005.138(1): 17-26.
[44] Jain M, Nijhawan A, Arora R, et al.F-box proteins in rice.Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress[J].Plant Physiol, 2007,143(4): 1467-1483.[45] Addo-Quaye C, Eshoo T W, Bartel D P, et al.Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome[J].Curr Biol, 2008,18(10): 758-762.
[46] German M A, Pillay M, Jeong D H, et al.Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends[J].Nat Biotechnol, 2008, 26(8): 941-946.
[47] Addo-Quaye C, Miller W, Axtell M J.CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets[J].Bioinformatics, 2009.25(1): 130-131.
[48] Liu Q P, Feng Y, Zhu Z J.Dicer-like (DCL) proteins in plants[J].Functional & Integrative Genomics, 2009.9(3): 277-286.
[49] Cho S H, Addo-Quaye C, Coruh C, et al.Physcomitrella patens DCL3 is required for 22-24 nt siRNA accumulation, suppression of retrotransposon-derived transcripts, and normal development[J].Plos Genet, 2008.4(12): e1000314.
[50] Fusaro A F, Matthew L, Smith N A,et al.RNA interferenceinducing hairpin RNAs in plants act through the viral defence pathway[J].Embo Rep, 2006.7(11): 1168-1175.
[51] Peng H, Chun J, Ai T B, et al.MicroRNA profiles and their control of male gametophyte development in rice[J].Plant Mol Biol, 2012, 80(1): 85-102.
Analysis Method for Small RNA Hhigh-throughput Sequenced Data
Peng Hua1,2, Li fosheng1, Wang Shenghua1
(1.College of Life Science, Sichuan University, Chengdu 610064, China; 2.Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China)
Here, we build a small RNA high-throughput sequenced data analysis platform by applying Perl language and MySQL database; through the analysis of four rice small RNA-Seq datasets, we introduce this method and dataf l ow in detail.We build a database for whole genome structure and known ncRNAs sites information based on the MSU 6.1 rice genome annotation.In combination with our perl script, the positioning and statistics of small RNAs can be achieved.Through the analysis of the known pre-miRNA, we extracte the feature of expression and designe a new algorithm used for new miRNAs discovery.A large number of new miRNAs can be fi ltered in this method, and 98% of known miRNAs can be fi ltered in the dataset.According to the diversity of rice small RNA, we also give a discussion and instruction about how to distinguish between endo-siRNAs and miRNAs in small RNA-Seq data.In this paper, a data analysis platform is designed, which is simple and efficient, and can be used as the storage and query media to analyze multiple hits reads for fl exible and diversif i ed statistical results.can be used for multiple hits reads analysis, can easily get more fl exible statistical results.According to this method, small RNA data analysis platforms of other model species can also be built.With the growing popularity of high-throughput sequencing in the future, the method will help small laboratories to establish their own data analysis platforms, which is of important and practical signif i cance.
small RNAs; non-coding RNA(ncRNAs); microRNAs(miRNAs); endo-siRNAs; small RNA high-throughput sequencing(small RNA-Seq)
Q531,Q751,TP391 [Document Code] A
10.11967/2017150204
Q531,Q751,TP391
A DOI:10.11967/ 2017150204
国家自然科学基金(31070276):植物不同启动子与雄配子体in-miRNA的表达关系
彭骅:博士研究生在读,研究方向:生物信息学,联系电话:010-64801262, 18510174449,联系邮箱:penghua@genetics.ac.cn李佛生:博士研究生在读,研究方向:植物生理与分子生物学,联系电话:028-85417281,联系邮箱:foshengli1987@163.com
王胜华:教授,研究方向:非编码RNA与植物生殖发育,联系电话:028-85417281,联系邮箱:wang_sh@scu.edu.cn
猜你喜欢
国际太空(2023年1期)2023-02-27
江苏科技信息(2022年16期)2022-07-17
今日农业(2021年11期)2021-08-13
透析与人工器官(2020年1期)2020-11-16
中西医结合肝病杂志(2020年2期)2020-10-27
铁道通信信号(2019年8期)2019-10-10
中成药(2018年7期)2018-08-04
中国发展观察(2017年8期)2017-04-26
图书馆建设(2015年10期)2015-02-13
新世纪图书馆(2014年7期)2014-09-19
