
功能性消化不良患者唾液菌群的研究*
2017-06-05 14:19刘婉薇李良芳岑荣英聂胜利卢艳娴李锐锋李子俊
重庆医学 2017年13期
陈 瑜,刘婉薇,李良芳,岑荣英,聂胜利,卢艳娴,,李锐锋,,李子俊,△
(1.南方医科大学,广州 510515;2.广东省人民医院/广东省医学科学院,广州 510080)
·生物信息学·
功能性消化不良患者唾液菌群的研究*
陈 瑜1,刘婉薇2,李良芳2,岑荣英2,聂胜利2,卢艳娴1,2,李锐锋1,2,李子俊1,2△
(1.南方医科大学,广州 510515;2.广东省人民医院/广东省医学科学院,广州 510080)
目的 分析功能性消化不良患者与健康对照者唾液菌群的差异。方法 收集功能性消化不良患者及健康对照者的唾液标本,提取总基因组DNA,采用高通量测序技术对样本中细菌的16S rRNA-V4区进行DNA测序后进行生物信息学分析。结果 功能性消化不良组唾液中的菌群以变形菌门为主,健康对照组以拟杆菌门为主。功能性消化不良唾液菌群的Chao1、ACE、Shannon指数分别为1 295、1 351、4.93,健康对照组为1 001、1 019、5.28。 PCoA可基本将两组间的唾液菌群区分开。LEfSe分析发现两组间有差异菌群共有16个,包括普式菌属、奈瑟菌属及变形菌门等。结论 功能性消化不良患者唾液有其特征性的菌群组成,以变形菌门为主,但两组间的丰富度及多样性无差别,奈瑟菌属是两组间差异显著的细菌之一。
功能性消化不良;唾液;细菌;奈瑟菌属
胃及食管长期以来均被认为“无菌”或仅有“过路菌”,而近年来通过高通量测序等技术发现胃及食管都有着其独特的细菌组成。Bik等[1]研究发现,胃黏膜中细菌主要由厚壁菌门及变形菌门组成,而细菌与上消化道疾病的关系也越来越受到重视。研究[2-3]发现,反流性食管炎、Barrett食管与正常的食管黏膜细菌组成不同,不同的细菌群落可能与食管疾病有关[4]。非萎缩性胃炎、肠化生、胃癌胃黏膜中的细菌多样性依次降低,非萎缩性胃炎与胃癌的细菌组成有显著不同[5]。此外,细菌与消化性溃疡、萎缩性胃炎也有密切联系。功能性消化不良(functional dyspepsia,FD)是一种常见的功能性胃肠病,其发病机制目前仍不清楚,包括胃排空延迟、容受性舒张功能下降、精神心理因素等[6-7]。胃与口腔及食管相通,口腔中的细菌可通过唾液吞咽等方式影响胃内细菌组成,有研究提示胃内的耐酸细菌主要是来源于口腔和食物的过路菌[8]。因此,笔者推测,唾液中的细菌改变与功能性消化不良的发生、发展有关。
1 资料与方法
1.1 研究对象 选择2015年7月至2016年3月广东省人民医院(广东省医学科学院)就诊的FD患者14例及同期行健康体检的健康对照者(health control,HC)10例作为研究对象。本研究已通过广东省人民医院(广东省医学科学院)的伦理委员会审查,所有研究对象均签署知情同意书。
1.2 纳入标准 FD患者需符合罗马Ⅲ诊断标准,健康对照者为无器质性疾病、系统性疾病、口腔疾病、肿瘤家族史的健康人,两组人群年龄均大于18岁,且无幽门螺杆菌(Helicobacter pylori,Hp)感染。
1.3 排除标准 近4周内有腹泻病史、感染性疾病史或急性胃肠炎病史;近4周内使用调节肠道菌群药物、抗生素、质子泵抑制剂、其他抑酸药物、非甾体类抗炎药及根除Hp药物等;有胃、食管及其他消化道手术史等; 合并其他部位肿瘤、器质性疾病、系统性疾病和急性口腔感染等;孕妇或哺乳期妇女。
1.4 方法
1.4.1 唾液收集 用50 mL无菌无酶离心管收集非刺激性唾液。唾液收集前需禁食、禁饮2 h以上,唾液总量5 mL以上,30 min内完成。收集完后1 h内放入-80℃冰箱保存。
1.4.2 唾液DNA提取 采用Ultra Clean Microbial DNA Isolation Kit提取唾液总DNA,具体步骤按说明书操作。
1.4.3 DNA检测 提取后的DNA需达以下标准:DNA总量大于或等于150 ng;OD 260/280值1.8~2.0,DNA浓度大于或等于5 ng/μL。样品检测合格后送至北京诺禾致源生物科技技术有限公司进行测序,测序平台为Illumina Hiseq PE250。
1.4.4 生物信息学处理 主要包括物种注释、操作分类单位(operational taxonomic unit,OTU)、 样品α多样性分析、β多样性分析等。
1.5 统计学处理 应用 SPSS21.0软件处理数据,年龄间差异及组间菌群相对丰度的差异采用Mann-WhitneyU检验,性别间差异采用χ2检验,OTU数量及α多样性指数差异采用两独立样本t检验。检验水准α=0.05,以P<0.05为差异有统计学意义。
2 结 果
2.1 一般情况 本研究纳入FD患者14例,健康对照者10例。FD平均年龄为35岁,健康对照组平均年龄41岁,年龄、性别比例组间比较差异无统计学意义(P>0.05)。
2.2 OTU分析及物种分析 FD和健康对照组的唾液样本共获得1 589 781条序列,平均每个样本为66 241条。FD组获得序列平均为66 675条,健康对照组获得序列平均为61 642条。以97%的一致性将序列聚类成为OTU,FD组平均得到(1 162±503)个OTU,健康对照组平均得到(928±425)个OTU。物种分析表明门水平的细菌有42种,属水平有309种,种水平有91种。 在门水平,FD组以变形菌门(Proteobacteria)为主,占40.7%,健康对照组以拟杆菌门(Bacteroidetes)为主,占37.3%。在属水平,FD组以奈瑟菌属(Neisseria)、梭杆菌属(Fusobacterium)、普式菌属(Prevotella)为主,健康对照组以普式菌属(Prevotella)、梭杆菌属(Fusobacterium)、韦荣球菌属(Veillonella)为主(图1)。

图1 两组唾液菌群门水平相对丰度柱形图
2.3 α多样性分析 当序列数达约30 000条,两组的稀释曲线达到平台期,表明测序深度已经基本覆盖大部分物种。两组的平均指数平均达到98%以上(图2)。FD的平均Chao1、ACE、Shannon及Simpson指数为1 295、1 351、4.93、0.90,健康对照组的为1 001、1 019、5.28、0.92,表明两组样本中细菌物种丰富且较为复杂。但上述指数组间比较差异均无统计学意义(P>0.05)。

图2 两组稀释曲线
2.4 β多样性分析 本试验采用Qiime软件,采用uniFrac进行β多样性分析。该方法可展示菌群之间的相似度[9]。两组菌群构成的主成分分析(Principal Co-ordinates Analysis,PCoA)显示,PCoA可基本将FD组和健康对照组唾液内细菌群落分开,说明两组细菌群落构成不同,差异有统计学意义(图3)。
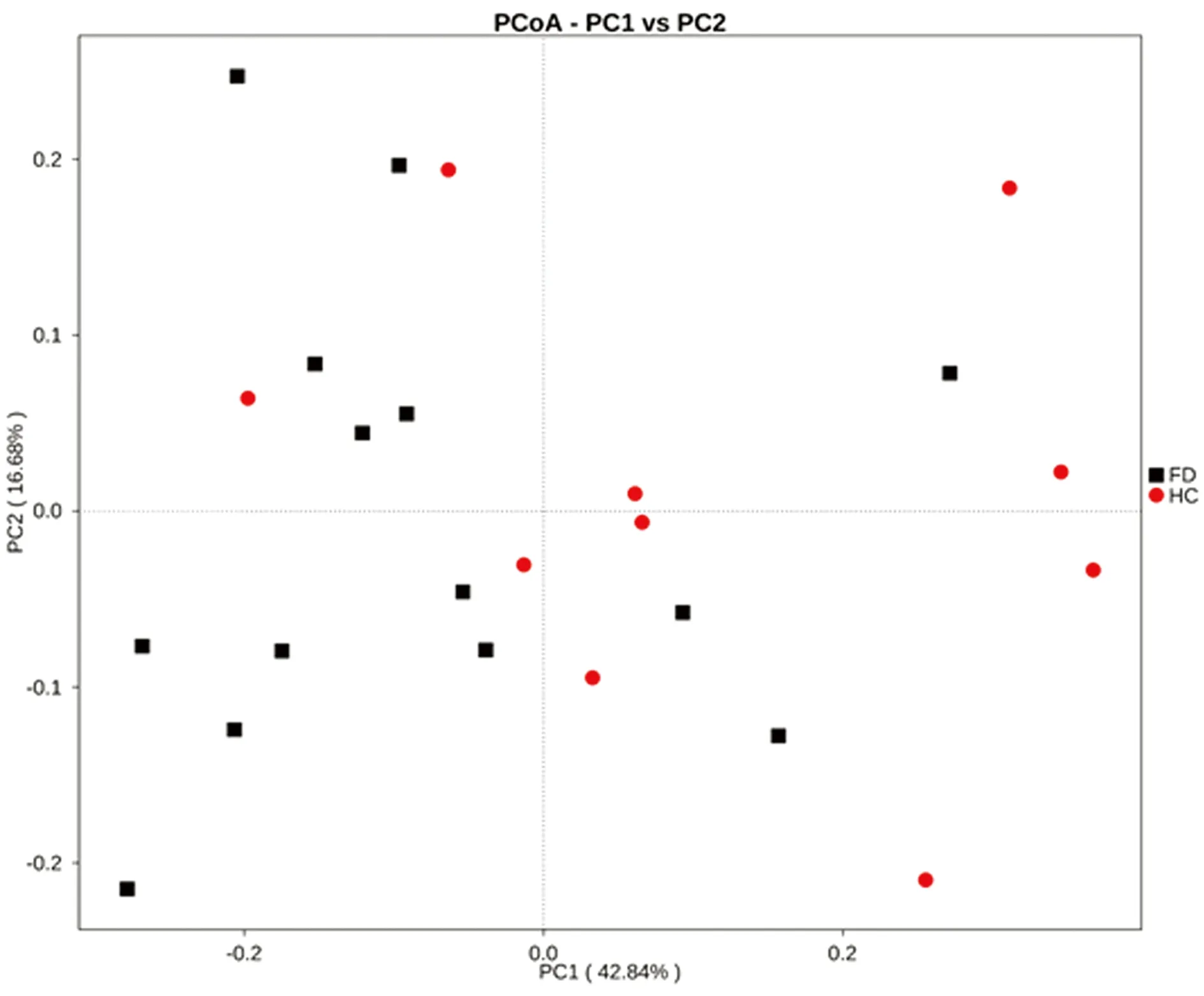
图3 功能性消化不良组和健康对照组唾液细菌群落构成PCoA分析
2.5 LEfSe分析 LEfSe是一种用于发现高维生物标识和揭示基因组特征的软件分析,能够在组与组之间寻找具有统计学差异的物种。LEfSe分析得出两组间有差异菌群共有16个,其中11个在健康对照组富集,分别为Bacteroidia、拟杆菌目(Bacteroidales)、拟杆菌门(Bacteroidetes)、普式菌属(Prevotella)、普雷沃菌科(Prevotellaceae)、厚壁菌门(Firmicutes)、梭状芽孢杆菌(Clostridia)、梭菌目(Clostridiales)、韦荣球菌科(Veillonellaceae)、韦荣球菌属(Veillonella)、dispar,5个在FD组富集,分别为奈瑟菌属(Neisseria)、奈瑟菌目(Neisseriales)、奈瑟菌科(Neisseriaceae)、β变形菌(Betaproteobacteria)及变形菌门(Proteobacteria)(图4)。
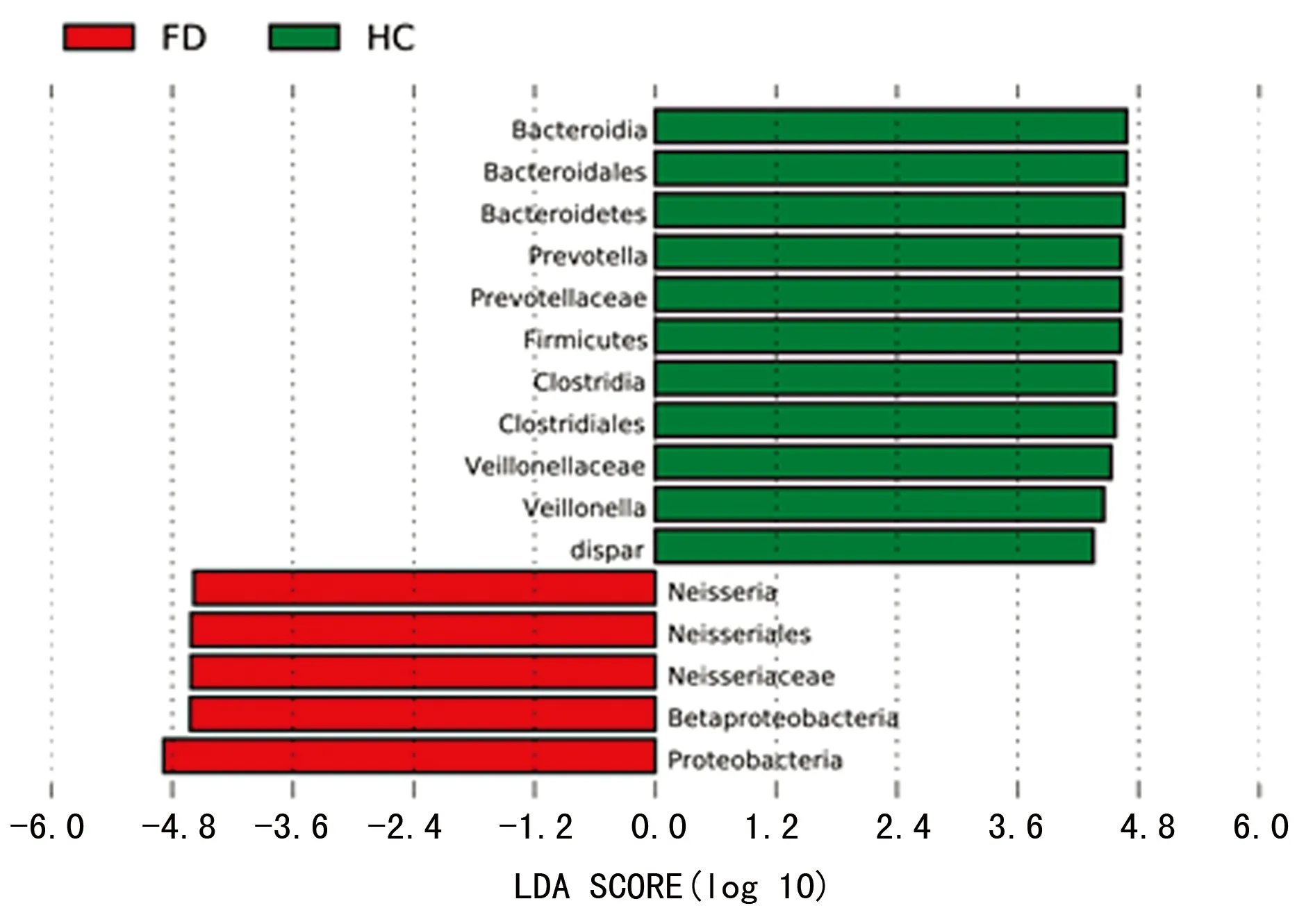
图4 LDA值分布柱状图
3 讨 论
细菌与上消化道疾病的发生发展关系密切。Blackett等[10]研究发现胃食管反流病及Barrett食管黏膜中弯曲杆菌属明显增加,且与IL-18增多相关,IL-18可介导体内免疫反应发生,可能与食管相关疾病发病有关。Khosravi等[11]发现消化性溃疡与链球菌属相关,可能是除Hp外另一个致病菌。Mattarelli等[12]发现胃酸过少患者的胃黏膜中存在大量双歧杆菌科,而正常黏膜中没有发现。此外,食管癌、胃癌等其他上消化道疾病也有其各自的微生物群落组成[13]。胃内的细菌组成部分来源于口腔,唾液是口腔微生态的介质之一,大量微生物存在于唾液中[14]。因此,研究唾液中的微生物组成,对上消化道疾病的发病、诊断、治疗等方面有重大意义。
本研究显示健康对照组唾液中以普式菌属、梭杆菌属、韦荣球菌属为主,与既往研究结果相似,而FD患者唾液中以奈瑟菌属、梭杆菌属、普式菌属为主。Alpha多样性分析包括稀释曲线、Chao1、ACE、Shannon等指数。 Chao1及ACE用于计算菌群的丰度,数值越大,说明物种总数越多。Shannon及Simpson用于计算菌群的多样性,Shannon值越大,Simpson值越小,说明群落多样性越高。这些结果结合稀释曲线、Coverage 指数及Rank Abundance曲线,表明两组测序深度足够,足以覆盖唾液中的大部分细菌种属,表明各组样品丰富度、多样性及均匀度良好。但上述指数的组间比较均无统计学意义,表明两组样品间丰富度及多样性无明显差异。PCoA分析表明两组间细菌群落构成不同,差异有统计学意义(P=0.008),FD患者唾液有其特征性的细菌组成。
LEfSe分析发现两组间有差异菌群共有16个,其中11个在健康对照组富集,其中大部分属于拟杆菌门(如普式菌属)及厚壁菌门(如韦荣球菌属),这二者在健康对照组门水平共占比约56.6%。另5个在FD组富集,分别为奈瑟菌属、奈瑟菌目、奈瑟菌科、β变形菌及变形菌门。这5种细菌均属于变形菌门,而变形菌门是功能性消化不良组在门水平占比最高的细菌,奈瑟菌属是属水平占比最高的细菌。
奈瑟菌属与消化道疾病关系密切。有研究发现,Hp阳性的上消化道疾病患者(包括胃溃疡、非溃疡性消化不良、反流性食管炎等)胃黏膜中除Hp外,主要的细菌为奈瑟菌属、链球菌属、罗氏菌属及葡萄球菌属,并且,非溃疡性消化不良黏膜中上述细菌比胃溃疡黏膜要高(P<0.01),这提示了其他的细菌在非溃疡性消化不良发病中的致病作用。Muto等[14]研究发现,相比于口腔中的其他细菌,奈瑟菌属可以在加入乙醇的培养基中产生大量的乙醛。长期积累的乙醛可能影响上呼吸道及消化道的上皮细胞,导致肿瘤发生。另外,Väkeväinen等[15]发现胃中的胃酸缺乏时,某些细菌(主要为奈瑟菌属及罗氏菌属)产乙醛的量增多,而乙醛是已知的致癌物质,因此,菌群的改变可能与胃酸缺乏的人上消化道的肿瘤发生有关。
胃内的细菌组成部分来源于口腔,奈瑟菌属属于人体口腔内的正常菌群,一般认为是无害的,但在特殊情况下(如长期酗酒)也可能对人体产生不利的影响。尽管功能性消化不良是一种功能性疾病,但这对指导患者的生活习惯也有一定的意义。此外,某些患者可能长期服用抗酸药/抑酸药[16],可能导致胃酸缺乏,因此,这对临床用药也有一定的指导意义。
本试验发现FD患者的唾液菌群结构与健康人不同,有其特征性的细菌组成,以变形菌门为主。并且,奈瑟菌属在FD唾液属水平中占比最高,且为两组间有差异性的细菌之一,其对FD的发病及治疗等方面可能有重大意义。但此研究尚存在许多不足之处。首先,样本量较少,有待扩大样本量进一步证实本研究所得结论。其次,未将功能性消化不良进行分型后检测其各自菌群变化,可在后续研究中继续细化,这对临床的诊断及治疗等方面更有意义。
[1]Bik EM,Eckburg PB,Gill SR,et al.Molecular analysis of the bacterial microbiota in the human stomach[J].Proc Natl Acad Sci U S A,2006,103(3):732-737.
[2]Nardone G,Compare D.The human gastric microbiota:Is it time to rethink the pathogenesis of stomach diseases?[J].United European Gastroenterol J,2015,3(3):255-260.
[3]Schulz C,Schütte K,Malfertheiner P.Helicobacter pylori and other gastric microbiota in gastroduodenal pathologies[J].Dig Dis,2016,34(3):210-216.
[4]Liu N,Ando T,Ishiguro K,et al.Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett′s esophagus[J].BMC Infect Dis,2013(13):130.
[5]Aviles-Jimenez F,Vazquez-Jimenez F,Medrano-Guzman R,et al.Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer[J].Sci Rep,2014(26):4202.
[6]杨昌妮,刘纯伦.精神心理因素与功能性消化不良的相关性研究进展[J].重庆医学,2015,44(15):2129-2131.
[7]王子恺,杨云生.胃内菌群与胃部疾病关系研究现状及展望[J].中华消化杂志,2014,34(3):210-211.
[8]Hamady M,Lozupone C,Knight R.Fast UniFrac:facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and Phylo Chip Data[J].ISME J,2010,4(1):17-27.
[9]Segata N,Izard J,Waldron L,et al.Metagenomic biomarker discovery and explanation[J].Genome Biol,2011,12(6):R60.
[10]Blackett KL,Siddhi SS,Cleary S,et al.Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease,Barrett′s and oesophageal carcinoma:association or causality?[J].Aliment Pharmacol Ther,2013,37(11):1084-1092.
[11]Khosravi Y,Dieye Y,Poh BH,et al.Culturable bacterial microbiota of the stomach of Helicobacter pylori positive and negative gastric disease patients[J].Scientific World J,2014:610421.
[12] Mattarelli P,Brandi G,Calabrese C,et al.Occurrence of bifidobacteriaceae in human hypochlorhydria stomach [J].Microb Ecol Health Dis,2014,25(9):5.
[13]Dicksved J,Lindberg M,Rosenquist M,et al.Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls[J].J Med Microbiol,2009,58(4):509-516.
[14]Muto M,Hitomi Y,Ohtsu A,et al.Acetaldehyde production by non-pathogenic Neisseria in human oral microflora:implications for carcinogenesis in upper aerodigestive tract[J].Int J Cancer,2000,88(3):342-350.
[15]Väkeväinen S,Tillonen J,Blom M,et al.Acetaldehyde production and other ADH-related characteristics of aerobic bacteria isolated from hypochlorhydric human stomach[J].Alcohol Clin Exp Res,2001,25(3):421-426.
[16]王洪波,李永芳,陈清波,等.兰索拉唑联合莫沙必利治疗功能性消化不良的疗效观察[J].临床内科杂志,2015,32(8):558-559.
Analysis on saliva microbiome in patients with functional dyspepsia*
Chen Yu1,Liu Wanwei2,Li Liangfang2,Cen Rongying2,Nie Shengli2,Lu Yanxian1,2,Li Ruifeng1,2,Li Zijun1,2△
(1.Southern Medical Universtiy,Guangzhou,Guangdong 510515,China;2.Guangdong Provincial People′s Hospital/Guangdong Academy of Medical Sciences ,Guangzhou,Guangdong 510080,China)
[Abstract] Objective To analyze the difference of salivary microbiome between the patients with functional dyspepsia and healthy controls.Methods Saliva samples were collected from the patients with functional dyspepsia and healthy control.Genomic DNA of the samples was extracted,and the high-throughput sequencing technology was used to conduct DNA sequence of 16S rRNA-V4 region.Subsequently,all the data were performed by the bioinformatic analysis.Results The salivary microbiome in the functional dyspepsia group was dominated by Proteobacteria,while Bacteroidetes was the top microbiota in the heathy control group.In the functional dyspepsia group,the Chao1 index,ACE index,Shannon index and Simpson index were 1 295,1 351,4.93 and 0.90 respectively.In the healthy control group,the above indexes were 1 001,1 351,5.28 and 0.92 respectively.The PCoA basically separated the microbiome composition of the two groups.Sixteen kinds of microbiota were significantly different between two groups using linear discriminant analysis effect size tool,including Bacteroidetes,Prevotella,Prevotellaceae,Neisseria,Betaproteobacteria and Proteobacteria,etc.Conclusion Saliva in the patients with functional dyspesia has characteristic microbiome composition,which is dominated by Proteobacteria,but the richness and diversity between the two groups have no difference.Neisseria is one of the significantly different bacteria between the two groups.
functional dyspepsia;saliva;microbiome;Neisseria
10.3969/j.issn.1671-8348.2017.13.020
广东省省级科技计划项目(2013B021800198)。 作者简介:陈瑜(1992-),硕士,主要从事功能性胃肠病与微生态研究。△
,E-mail:zijunli2005@aliyun.com。
R573.5
A
1671-8348(2017)13-1789-03
2016-12-07
2017-01-25)
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
世界科学技术-中医药现代化(2022年3期)2022-08-22
中国生殖健康(2020年8期)2021-01-18
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
模具制造(2019年3期)2019-06-06
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
中国生殖健康(2018年3期)2018-11-06
读者·校园版(2017年9期)2017-04-15
