
参与肝纤维化形成的细胞和细胞因子的新进展
2017-06-05 15:13:28梁文杰杜雅菊
胃肠病学和肝病学杂志 2017年5期
梁文杰, 陈 晶, 杜雅菊
哈尔滨医科大学附属第二医院消化内科,黑龙江 哈尔滨 150086
参与肝纤维化形成的细胞和细胞因子的新进展
梁文杰, 陈 晶, 杜雅菊
哈尔滨医科大学附属第二医院消化内科,黑龙江 哈尔滨 150086
肝纤维化是各种慢性肝病的共同病理过程,许多类型的细胞与细胞因子参与肝纤维化的发生、发展。最近,对参与肝纤维化的细胞和细胞因子分子机制有了新的认识。终止肝纤维化的过程,可能会逆转晚期肝纤维化甚至肝硬化。很多研究已更好地阐述肝纤维化所涉及的细胞和细胞因子分子机制。肝星状细胞的激活是肝纤维化的中心事件,辅以其他基质来源的细胞。确定不同类型细胞的相互作用,揭示细胞因子对这些细胞的调节机制,会发现新的治疗靶点。本文就细胞及细胞因子在肝纤维化发病中的作用机制作一概述。
肝纤维化;细胞;细胞因子
肝脏疾病及随后的肝功能异常、肝衰竭是一个巨大的临床挑战。在美国,肝衰竭所致死亡位居第12位,在中年人中死亡人数跃居第4位[1]。肝癌风险率逐渐增高的原因包括非酒精性脂肪肝的发病率增高、丙型肝炎疫苗的缺乏和人口的老龄化。肝移植是肝硬化、肝功能衰竭的主要治疗方法,是直接改变肝脏疾病所致死亡的唯一治疗方法。但肝脏疾病发病率高,肝移植成本高,需要终身免疫抑制治疗,且器官供体短缺,不能满足肝硬化患者的需求。了解参与肝纤维化形成的细胞及细胞因子机制,为改善这种慢性疾病提供理论依据。
1 参与肝纤维化的细胞
肝脏由实质细胞(肝细胞)和非实质细胞构成。肝血窦壁主要由肝星状细胞(hepatic stellate cells, HSCs)、肝窦内皮细胞(hepatic sinusoidal endothelial cells, HSEC)、Kupffer细胞组成。这些细胞均通过直接或间接作用参与HSCs的激活,进而参与肝纤维化的形成。
1.1 HSCs HSCs占正常肝脏的5%~8%,又称贮脂细胞、Ito细胞、窦周细胞,或富含维生素A的细胞。主要功能是贮存维生素A和其他类维甲酸;细胞外基质(extracellular matrix, ECM)的合成与降解;调节肝窦血流[2]。各种创伤因子,如肝细胞凋亡小体和Kupffer细胞分泌的肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)、转化生长因子-β(transforming growth factor-β)和转化生长因子-α(TGF-α)、活性氧(reactive oxygen species, ROS),可促进HSCs的活化。活化状态的HSCs具有肌成纤维母细胞(myofibroblasts, MFs)的特点,使具有收缩功能的蛋白质表达增加(如:α-平滑肌激动蛋白、平滑肌肌球蛋白);同时,促进肝纤维化发生的相关受体表达增加(如:促纤维化因子、趋化因子和促有丝分裂因子的受体),产生、分泌ECM和胶原蛋白。ECM的合成与降解受基质金属蛋白酶(matrix metalloproteinases, MMPs)和基质金属蛋白酶抑制(inhibitors of the metalloproteinase, TIMPs)调节。至今对于MFs的来源仍有争议。因在肝损伤时,ECM主要的来源细胞是HSCs,所以HSCs被认为是MFs来源的主要细胞[3]。此外,MFs也来源于骨髓间充质细胞、上皮-间质转化细胞(epithelial-mesenchymal transition,EMT)及内皮-间质转化细胞[4]。
1.2 Kupffer细胞 Kupffer细胞约占肝脏非实质细胞的20%~25%。在肝脏中的主要作用是清除门脉循环中的异物。在肝损伤时,Kupffer细胞活化,合成、分泌促纤维化细胞因子(如TNF-α、血小板生长因子、TGF-β),激活HSCs[5]。TNF-α作用于肝细胞,通过氧化应激和细胞凋亡使肝损伤[6]。 活化的Kupffer细胞可释放炎性因子(如IL-6)促进炎性反应;也可释放趋化因子,包括CXC亚家族(如 CXCL1、CXCL2、CXCL8和 CXCL16)。其中CXCL1、CXCL2、CXCL8通过结合趋化因子受体CXCR1和CXCR2,从而募集中性粒细胞,释放细胞毒素和ROS,促进肝细胞损伤和肝纤维化的发展[7]。但在肝脏损伤恢复期,Kupffer细胞也可释放保护性炎性因子,如IL-10, IL-10可通过Stat3信号上调细胞转导抑制因子3(SOCS3)的表达[8],使SOCS3与gp130结合[9],从而达到抑制炎症反应的目的。
1.3 HSEC HSEC是构成肝窦壁的主要细胞,占肝脏非实质细胞的44%~60%。HSEC的特性是具有窗口,缺乏典型的基底膜。HSEC窗口测量直径为150~175 nm,是一种动态过滤器,利于肝血窦与肝实质细胞进行物质交换[10]。在肝损伤时,HSEC失窗口化,合成TGF-β或血小板生长因子-BB(platelet-derived growth factor-BB, PDGF-BB)分泌ECM,导致HSCs激活[11]。同时,在HSEC中,NO合成酶(eNOS)活性下降,使肝内NO合成减少,进而导致肝内血管收缩,导致门脉高压。此外,HSEC也会导致血管收缩物质增加(如:TXA2),从而增加肝内血管阻力,加重肝纤维化[12]。
1.4 肝细胞 肝细胞是肝脏的实质细胞,在肝纤维化中发挥重要的作用。在肝损伤时,肝细胞凋亡发生在早期阶段,肝细胞凋亡小体可促进Kupffer细胞分泌促炎因子与促纤维化因子,也可直接激活HSCs。其次是肝细胞增殖,IL-6通过Stat3促进肝细胞由G1期到S期的循环发展[13],同时也可作用于其他的抗凋亡基因(包括FLIP、Bcl-2和Bcl-xL)[14-15],促进肝细胞再生与有丝分裂。此外,在急性肝损伤时,肝细胞也可通过TGF-β-Smad2/3信号通路参与EMT[16],成为MFs的来源之一,参与肝纤维化的形成。
2 参与肝纤维化的细胞因子
除多种细胞参与肝纤维化的过程,TGF-β、血管内皮生长因子(vascular endothelial growth factor, VEGF)、PDGF、IL、内皮素(endothelin, ET)等细胞因子在肝纤维化的过程中也起关键作用。
2.1 TGF-β TGF-β由Kupffer细胞、巨噬细胞、HSEC、肝细胞和HSCs产生。其有3种主要形式:TGF-β1、TGF-β2、TGF-β3。其中,TGF-β1是肝脏疾病中主要的促纤维化因子,TGF-β1受兴奋性信号(Smad 2和3)和抑制性信号(Smad 7)调节。TGF-β1致肝纤维化可通过以下几个机制:(1) TGF-β1在HSCs活化为MFs中起重要作用。活化的HSCs,也可促进TGF-β1产生[17]。(2) HSCs中TGF-β1通过调节MMPs、血浆纤溶酶原活化抑制因子-1(plasminogen activator inhibitor-1, PAI-1)和TIMPs使细胞外基质成分增加[18]。主要刺激Ι型胶原的产生,但对其他细胞外基质成分(如细胞纤连蛋白、蛋白聚糖)的产生也有刺激作用[19]。(3) TGF-β1已被证明能够抑制DNA的合成和诱导肝细胞凋亡[20]。
2.2 VEGF VEGF是一种多功能蛋白,作用于其受体VEGFR-1和VEGFR-2,刺激血管内皮细胞增殖、迁移和调节血管渗透性。在肝损伤时,VEGF通过促进炎症反应、增加HSEC释放纤维增强因子和直接作用于HSCs,促进肝纤维化[21]。在HSEC中,VEGF即可诱导新生血管形成,也可通过激活Akt(丝氨酸/苏氨酸激酶)信号通路[22-23]增加血管张力;同时,通过eNOS产生NO,增加血管内的血流量[24],进而导致门脉高压的形成,加重肝纤维化。
2.3 PDGF PDGF家族包括5个二聚体蛋白:PDGF-AA、PDGF-BB、PDGF-CC、PDGF-DD和PDGF-AB。PDGF是HSCs增殖最强的有丝分裂原。在肝损伤时,PDGF可通过磷脂酰肌醇3激酶(PI3K)-Akt途径促进HSCs的活化。同时,也可通过跨膜受体酪氨酸激酶启动多个信号通路[25],包括丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)信号通路、细胞外信号调节酶(extracellular signal-Regulated protein kinase, EPK)信号通路及应激活化蛋白激酶信号通路(stress-activated protein kinase, SAPK),促进HSC的增殖。
2.4 IL IL是由炎性细胞产生的细胞因子,其参与肝纤维化的发生是相对复杂的。IL-1可直接激活HSCs,并刺激它产生MMP-9、MMP-13和TIMP-1,导致肝纤维化。在缺乏IL-1受体的小鼠中,可以减轻肝损伤和肝纤维化[26]。IL-17也可促进肝纤维化,其促进肝纤维化主要以下两种机制:(1)IL-17直接刺激HSCs产生Ⅰ型胶原蛋白,并通过Stat3信号通路促进它们活化为MFs[27]。(2)IL-17也可刺激Kupffer细胞表达IL-6、IL-1β、TNF-α炎性因子和纤维化发生的主要细胞因子TGF-β1。
此外,也有一些IL可抑制肝纤维化的发生[11]。IL-10是一种细胞因子,抑制炎症反应,对肝纤维化有调节作用[28]。同时,Huang等[29]在小鼠实验模型中证实,小鼠IL-10基因可通过抑制HSCs的活化和促进胶原蛋白的降解,抑制肝纤维化的发生。在小鼠模型中已证实,IL-22 通过抑制HSCs衰老,抑制肝纤维化,促进肝纤维化的修复[30]。
2.5 ET ET是由21种氨基酸组成的多肽,主要由ET-1、ET-2 和 ET -3三种异构体,以ET-1的血管作用最强。在肝损伤时,ET-1的产生增加和细胞来源发生转变,由内皮细胞转变为HSCs。ET-1和内皮素受体 A (endothelin receptor A, ETRA)结合后可上调 P-Akt表达水平,形成 ET-1-ETRA-P13K-Akt 链,促进HSC收缩、增殖和ECM的合成,从而促进肝纤维化的发生[31]。同时,ET-1也可直接收缩肝内血管,导致门脉高压症的发生。
3 肝纤维化的形成
在酒精、病毒、自身免疫等原因导致的肝纤维化中,HSC的激活是肝纤维化发生的中心环节。多种细胞及细胞因子通过复杂的途径参与肝纤维化的形成(见图1)。Kupffer细胞、肝细胞可直接激活HSC,也可释放促纤维化因子(如:TGF、PDGF)和炎性因子(如IL)通过复杂的细胞分子信号促进HSC的活化及促进炎症反应,加重肝纤维化。HSEC是构成肝血窦壁的主要细胞,是肝纤维化形成的血管因素的重要细胞。HSEC除可释放促纤维化因子和炎性因子,在增加肝内血管阻力导致门脉高压症中也起重要的作用。VEGF、ET通过相应的分子机制诱导血管再生和肝内血管收缩,从而导致门脉高压的形成,加重肝纤维化。其他细胞与细胞因子在肝纤维化的形成中也有一定的作用,如T细胞、NK细胞、表皮生长因子(epidermal growth factor,EGF)、肝细胞生长因子(hepatocyte growth factor,HGF)、干扰素(Interferon,IFN)等。
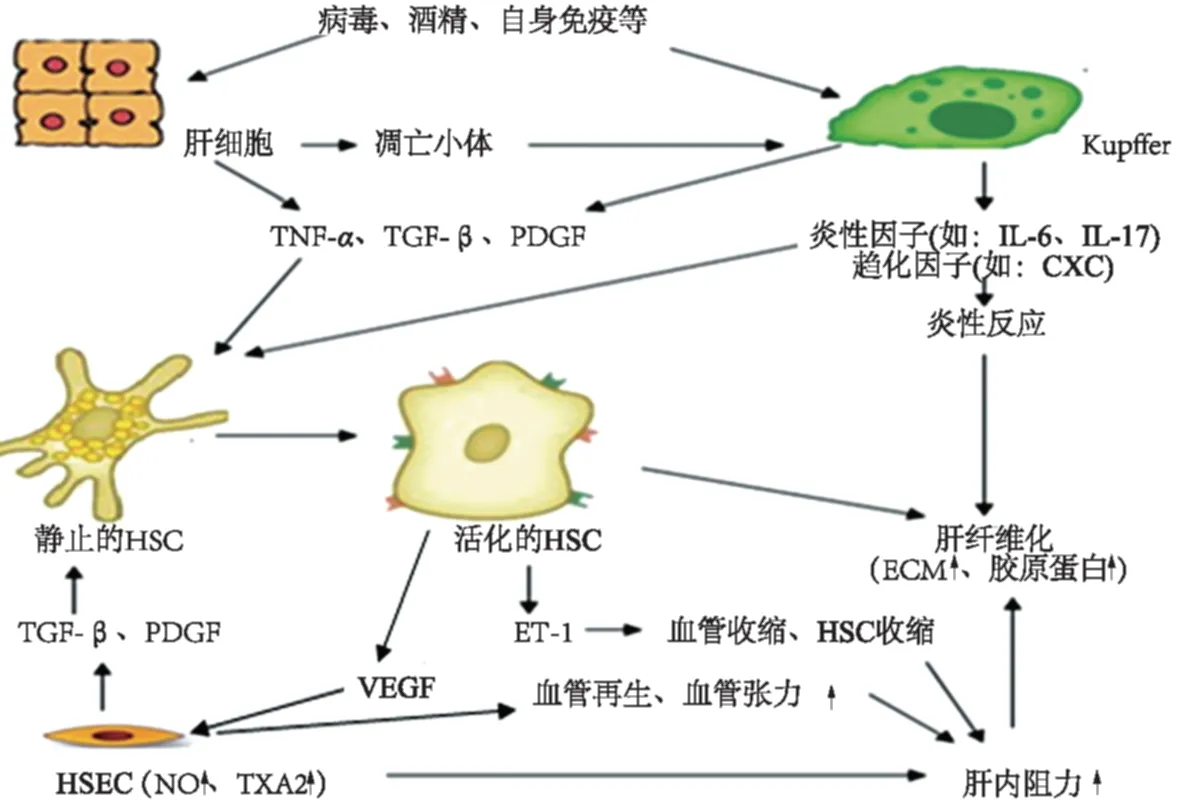
图1 细胞与细胞因子参与肝纤维化的分子途径Fig 1 Molecular pathways of cells and cytokines in liver fibrosis
总之,目前对肝纤维化形成机制的理解有了相当大的进步。ECM的增加,是肝纤维化形成过程中的关键因素。细胞外基质,星状细胞、内皮细胞和免疫细胞之间的相互作用已被证实。肝纤维化发生、发展过程相对复杂,从而为肝纤维化的治疗提供多个潜在靶点。虽然目前还没有具体的、安全的、有效的抗肝纤维化治疗方法,一旦开发出作用于上述细胞与细胞因子的药物,它们将影响肝纤维化治疗的进展。
[1]Bhatia SN, Underhill GH, Zaret KS, et al. Cell and tissue engineering for liver disease [J]. Sci Transl Med, 2014, 6 (245): 245sr2.
[2]Su TH, Kao JH, Liu CJ. Molecular mechanism and treatment of viral hepatitis-related liver fibrosis [J]. Int J Mol Sci, 2014, 15(6): 10579-10581.
[3]Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis [J]. Best Pract Res Clin Gastroenterol, 2011, 25(2): 305-317.
[4]Elpek GÖ. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update [J]. World J Gastroenterol, 2014, 20(23): 7260-7276.
[5]Seki E, Tsutsui H, Nakano H, et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1β [J]. J Immunol, 2001, 166(4): 2651-2657.
[6]Suraweera DB, Weeratunga AN, Hu RW, et al. Alcoholic hepatitis: The pivotal role of Kupffer cells [J]. World J Gastrointest Pathophysiol, 2015, 6(4): 90-98.
[7]肖春阳, 陆伦根. 免疫细胞在肝纤维化发生发展中的作用[J]. 临床肝胆病杂志, 2015, 31(9): 1532-1536. Xiao CY, Lu LG. Research advances in immune cellular pathogenesis in liver fibrosis [J]. J Clin Hepatol, 2015, 31(9): 1532-1536.
[8]Yasukawa H, Ohishi M, Mori H, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages [J]. Nat Immunol, 2003, 4(6): 551-556.
[9]Wang H, Lafdil F, Kong X, et al. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target [J]. Int J Biol Sci, 2011, 7(5): 536-550.
[10]Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis [J]. World J Gastroenterol, 2014, 20(23): 7312-7324.
[11]Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis [J]. Am J Physiol Cell Physiol, 2013, 305(8): C789-C799.
[12]Iwakiri Y. Pathophysiology of portal hypertension [J]. Clin Liver Dis, 2014, 18(2): 2-3.
[13]Terui K, Ozaki M. The role of STAT3 in liver regeneration [J]. Drugs Today (Barc), 2005, 41(7): 461-469.
[14]Fujiyoshi M, Ozaki M. Molecular mechanisms of liver regeneration and protection for treatment of liver dysfunction and diseases [J]. J Hepatobiliary Pancreat Sci, 2011, 18(1): 13-22.
[15]Kovalovich K, Li W, DeAngelis R, et al. Interleukin-6 protects against fas-mediated death by establishing a critical level of antiapoptotic hepatic proteins FLIP, Bcl-2, and Bcl-Xl [J]. J Biol Chem, 2001, 276(28): 26605 -26613.
[16]Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition [J]. J Biol Chem, 2007, 282(32): 23337-23347.
[17]Cong M, Iwaisako K, Jiang CY, et al. Cell signals influencing hepatic fibrosis [J]. Int J Hepatol, 2012, 2012: 158547.
[18]Pereira TN, Walsh MJ, Lewindon PJ, et al. Paediatric cholestatic liver disease: Diagnosis, assessment of disease progression and mechanisms of fibrogenesis [J]. World J Gastrointest Pathophysiol, 2010, 1(2): 73-79.
[19]Yoshida K, Matsuzaki K. Differential regulation of TGF-β/Smad signaling in hepatic stellate cells between acute and chronic liver injuries [J]. Front Physiol, 2012, 3: 53.
[20]Kirmaz C, Terzioglu E, Topalak O, et al. Serum transforming growth factor-beta1 (TGF-beta1) in patients with cirrhosis, chronic hepatitis B and chronic hepatitis C [J]. Eur Cytokine Netw, 2004, 15(2): 112-116.
[21]Liu Y, Kwon J, Popov Y, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice [J]. Gastroenterology, 2014, 146(5): 1339-1350.
[22]Fernandez M, Mejias M, Angermayr B, et al. Inhibition of VEGF receptor-2 decreases the development of hyperdynamic splanchnic circulation and portal-systemic collateral vessels in portal hypertensive rats [J]. J Hepatol, 2005, 43(1): 98-103.
[23]Abraldes JG, Iwakiri Y, Loureiro-Silva M, et al. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the intestinal microcirculatory bed, leading to a hyperdynamic state [J]. Am J Physiol Gastrointest Liver Physiol, 2006, 290(5): G980-G987.
[24]Huang HC, Haq O, Utsumi T, et al. Intestinal and plasma VEGF levels in cirrhosis: the role of portal pressure [J]. J Cell Mol Med, 2012, 16(5): 1125-1126.
[25]Pinzani M, MacIas-Barragan J. Update on the pathophysiology of liver fibrosis [J]. Expert Rev Gastroenterol Hepatol, 2010, 4(4): 459-472.
[26]Gieling RG, Wallace K, Han YP. Interleukin-1 participates in the progression from liver injury to fibrosis [J]. Am J Physiol Gastrointest Liver Physiol, 2009, 296(6): G1324-G1331.
[27]Meng FL, Wang K, Aoyama T, et al. IL-17 signaling in inflammatory cells, Kupffer cells and Hepatic Stellate cells exacerbates liver fibrosis [J]. Gastroenterology, 2012, 143(3): 765-776, e1-e3.
[28]Yue-Hong H, Yun-Xin C, Li-Juan ZH, et al. Hydrodynamics-based transfection of rat interleukin-10 gene attenuates porcine serum-induced liver fibrosis in rats by inhibiting the activation of hepatic stellate cells [J]. Int J Mol Med, 2014, 34(3): 677-686.
[29]Huang ZH, Chen YX, Zhang LJ, et al. Hydrodynamics-based transfection of rat interleukin-10 gene attenuates porcine serum-induced liver fibrosis in rats by inhibiting the activation of hepatic stellate cells [J]. Inte J Mol Med, 2014, 34(3): 677-686.
[30]Kong X, Feng D, Wang H, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice [J]. Hepatology, 2012, 56(3): 1150-1159.
[31]Khimji AI, Rockey DC. Endothelin and hepatic wound healing [J]. Pharmacol Res, 2011, 63(6): 512-518.
(责任编辑:王全楚)
New progress of the cells and cytokines involved in the formation of liver fibrosis
LIANG Wenjie, CHEN Jing, DU Yaju
Department of Gastroenterology, the Second Affiliated Hospital of Harbin Medical University, Harbin 150086, China
Liver fibrosis is a common pathological process of various chronic liver diseases. Many types of cells and cytokines are involved in the occurrence and development of liver fibrosis. Recently, a new understanding of the cellular and molecular mechanisms of liver fibrosis occurrs. Data indicate that the termination of fibrogenic processes may allow the reversal of advanced fibrosis and even cirrhosis. A lot of researches can better explain the cellular and molecular mechanisms involved in liver fibrosis. Activation of hepatic stellate cells remains a central event in fibrosis, complemented by other sources of matrix-producing cells. To determine the interaction of different types of cells, to reveal the role of cytokines on these cells and regulatory mechanisms, will find new therapeutic targets. In this paper, the pathogenesis of liver fibrosis from the aspects of cells and cytokines were summarized.
Liver fibrosis; Cell; Cytokine
10.3969/j.issn.1006-5709.2017.05.028
综述
梁文杰,在读硕士研究生,研究方向:肝纤维化的发病机制。E-mail:1406416522@qq.com
杜雅菊,主任医师,硕士研究生导师,教授,研究方向:胃肠病与肝脏疾病。E-mail:duyaju1964@163.com
R575
A
1006-5709(2017)05-0581-04
2016-07-04
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
昆明医科大学学报(2022年2期)2022-03-29 00:52:18
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
癌变·畸变·突变(2016年3期)2016-02-27 06:15:36
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
哈尔滨医药(2015年4期)2015-12-01 03:57:54
西南国防医药(2015年11期)2015-02-28 19:38:46
疑难病杂志(2014年12期)2014-04-16 05:19:29
无机化学学报(2014年10期)2014-02-28 17:33:13
