
An efficient green route for hexamethylene-1,6-diisocyanate synthesis by thermal decomposition of hexamethylene-1,6-dicarbamate over Co3O4/ZSM-5 catalyst:An indirect utilization of CO2☆
2017-05-28 19:45MuhammadAmmarYanCaoPengHeLiguoWangJiaqiangChenHuiquanLi
Muhammad Ammar ,Yan Cao *,Peng He Liguo Wang Jiaqiang Chen Huiquan Li*
1 Key Laboratory of Green Process and Engineering,National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology,Institute of Process Engineering,CAS,Beijing 100190,China
2 University of Chinese Academy of Sciences,Beijing 100049,China
1.Introduction
HDI is one of the most widely used aliphatic diisocyanate because of its unique properties such as less reactive as compared to other diisocyanate,excellen theatresistance,high resistance to turning yellow and high demand in industries for various applications[1,2].Conventionally,HDI is produced by phosgenation of amine at industrial scale[3].Although phosgene route has reactivity and yield advantages over other routes,it is extremely toxic and poses an environmental hazard due to difficulty in handling of phosgene with seriously corrosive byproduct HCl[4].Thus,it is highly desired to explore and develop phosgene-free green route for the synthesis of HDI.There are a number of phosgene-free routes reported in the literature[4-7].However,these routes have a number of issues such as toxicity of raw materials prepared from toxic chemicals,production of the considerable amount of by-products along with desired products,difficult separation of products and also hard to scale-up for industrial application.Among the phosgene-free routes,the thermal decomposition of HDC not only seems the most promising way to obtain HDI but also is an indirect way of chemical utilization of greenhouse CO2gas.Since the raw material HDC is usually obtained from dimethyl carbonate(DMC)or methyl carbamate(MC),which is derived from CO2.But a relatively less report on this route is available in the literature[4,8].The thermal decomposition route for the synthesis of HDI involves two stages,HDC first decomposes to intermediate hexamethylene-1-carbamate-6-isocyanate(HMI)and then HMI decompose to HDI(Fig.1).However,most ofthis route uses high boiling point solvent which suffers from difficulty in temperature control;high temperature may result in the fragmentation of aliphatic carbamate,many side reactions of highly active and unstable isocyanate and reverse reaction.
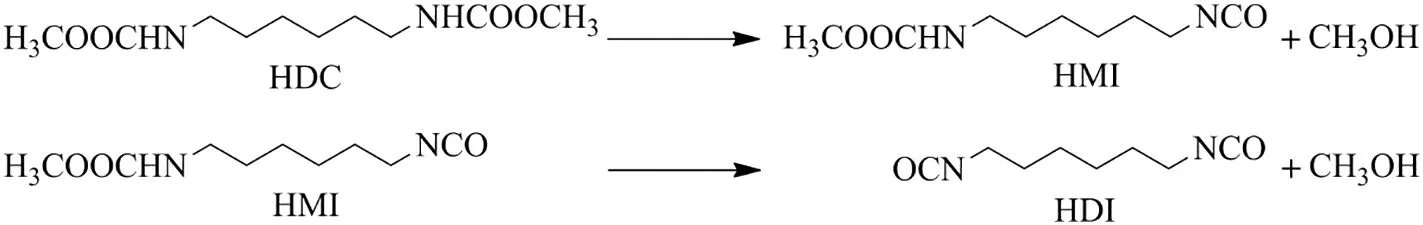
Fig.1.Thermal decomposition of HDC to HDI.
Recently,our group developed a new route for the thermal decomposition of carbamate to isocyanate using chlorobenzene as low boiling point solvent and achieved remarkable results from the thermal decomposition of methyl N-phenyl carbamate(MPC)to phenyl isocyanate(PI)without any catalyst[9].Compared to the routes using high boiling point solvent,this route reveals the bene fits of relatively low reaction temperature,low viscosity,high dispersion and avoidance of severe polymerization[10].However,without an effective catalyst,the efficiency of the thermal decomposition of HDC to HDI is limited when this new route is applied.This issue is expected to be overcome by proper catalyst utilization at relatively low temperature.
Several catalysts have been used in the phosgene-free route for the synthesis of HDI from thermal decomposition of HDC.Takahito et al.[11]used a homogenous di-n-butyltin oxide catalyst in the thermal decomposition of HDC to obtain HDI with selectivity of 82.4%at temperature of 230°C.Utilization of this homogeneous catalystcaused difficulty in separation and recovery,which limited its industrial application.Heterogeneous catalysts are secure,reasonable and environment-friendly.Furthermore the methodsusing heterogeneous catalystshave advantages for instance better selectivity,moderate reaction conditions,minimum waste and cheaper material of construction[12].However,very few heterogeneous catalysts for the decomposition of HDC to HDI have been reported.Serglo et al.[13]conducted thermal decomposition of HDC in decahydronaphthaleneas solvent,using montromrillonite K-10 as catalyst.A 97%HDC conversion was obtained after 24 h reaction time at 183°C.However,the authors did not report the HDI yield in that work.Da-Lei Sun et al.[14]used ZnAlPO4catalyst in the thermal decomposition of HDC to HDI,using dioctyl phthalate(DOP)as solvent.A 91.2%HDC conversion with 89.4%HDI yield was achieved at 350°C.Min Jeog Hyun et al.[15]developed phosgene-free decomposition of HDC over ZnO,using polyethylene glycol dimethyl ether(PGDE)as solvent.The HDC conversion reached 100%with 97%HDI yield after 2.5 h reaction time at 180°C.Although this route revealed high HDI yield,ZnO could not show good reusability due to ZnO leaching into the reaction mixture and structure transformation during recycling.However,the development in the heterogeneous catalysis for thermal decomposition of HDC is highly challenging.All the catalyst reported still does not meet the requirement of the industrialization because the reaction time is still too long and catalyst cannot be reused.To overcome this long-standing challenge,a new highly efficient and stable heterogeneous catalyst is needed.
Recently,many efforts have been made to develop an efficient heterogeneous catalyst by depositing transition metal oxides(ZnO,Cr2O3,Co3O4etc.)on a support such as ZSM-5,SBA-15,MCM-41 and so on[16-18].ZSM-5 is one of the most commonly used zeolites due to its thermal stability,unique channel structure,large surface,acidity and shape-selectivity.It has been widely used as the support in various moderate acid-catalyzed reactions[19].Modification of ZSM-5 with metaloxides has been reported to modify the ZSM-5 texturalproperties and affects the selectivity to hydrocarbon compounds[20-22].Furthermore,ZSM-5 can serve as host to activate metal ions,offering a unique ligand system with multiple types of coordination for cations.It was reported that the presence of other cations such as Na+and NH4+on the metal ion sitting caused deformation and an expected reconstruction of the Si-O-Al bridges could occur.Consequently,the acidity of the catalyst can be affected;thereby it may be expected to show higher potential in moderate acid-catalyzed decomposition reaction of HDC than metal oxides[23].However,no application of metal oxide supported ZSM-5 catalyst in the thermal decomposition of HDC has been realized so far.
In this work,metal oxide supported catalysts were used for the first time in the thermal decomposition of HDC to HDI using low boiling point solvent.Different metal oxide supported catalysts were prepared and screened for thermal decomposition of HDC.The physicochemical properties of most active Co3O4/ZSM-525catalyst were characterized by different techniques.The relationship between physicochemical properties and HDI selectivity was also explored to reveal the possible reason for high performance of Co3O4/ZSM-525catalyst.The reusability of the Co3O4/ZSM-525(PEG)catalyst and effects of reaction conditions were studied.Furthermore,a possible reaction mechanism was also proposed.Herein,we reportan indirectapproach to chemicalutilization of CO2by developing an efficient and environmentally friendly green route for thermal decomposition of HDC to HDI.
2.Experimental
2.1.Materials
The ZSM-5 zeolites(SiO2/Al2O3=25,38,60 and 170),SBA-15,and MCM-41 were purchased from the Catalyst Plant of Nankai University and XFNANO Advance Material Supplier,and used as support.Zinc nitrate hexahydrate(98.5%),Cobalt nitrate hexahydrate(98.5%),Chlorobenzene(≥99.0%),Methanol(≥99.0%)and Biphenyl were purchased from Sinopharm Chemical Reagent Co.,Ltd.,China.Polyethylene glycol PEG(MW=400)and Chromium nitrate nanohydrate(99.0%)were purchased from Aladdin Chemical Co.,Ltd.,Shanghai,China.Ammonia aqueous solution(28%)was purchased from Xilong Chemical Co.,Ltd.,China.HDI(>98%)was purchased from Tokyo Chemical Industry Co.,Ltd.,Japan.All these reagents were used as received without further purification.HDC was synthesized by the reaction of HDI with methanol(m(HDI):m(Methanol)=1:5)at 35°C for 12 h.HDC(>99%)was finally obtained by purification and confirmed by Mass Spectrometry and FTIR Spectroscopy.HMI was obtained in the form of HDC,HMI and HDI mixture,by the reaction of HDI with methanol(m(HDI):m(Methanol)=3:1)at 35°C for 1.5 h.,since HMI was not commercially available.
2.2.Preparation of catalysts
The catalysts were prepared by incipient wetness impregnation(IWI)method,PEG-additive(PEG)method and deposition precipitation with ammonia evaporation(DP)method in this study.Prior to metal loading,ZSM-5 zeolites were calcined at 500°C for 3 h under static air in a muf fle furnace.The theoretical loading of metal was 20 wt%in all the catalysts.The IWI catalyst was prepared by mixing 3 g of support(ZSM-5,SBA-15 or MCM-41)with an aqueous solution of corresponding precursor(Co(NO3)2·6H2O,Zn(NO3)2·6H2O or Cr(NO3)3·9H2O)to give 20 wt%of metal/support catalyst.The volume of solution necessary to completely wet the support was used.Then,the catalyst was sonicated in an ultrasonic bath at 30°C for 1 h[23].The PEG catalyst was prepared by adding Co(NO3)2·6H2O(3.741 g)and PEG(MW=400,4.714 g)into a solvent containing methanol(30 ml)and deionized water(90 ml).The resulting solution was re fluxed at 90°C for 2 h.3 g of ZSM-5 support was added into the solution subsequently.The obtained suspension was heated at 100°C with continuous stirring to evaporate the solvent[24].The DP catalyst was prepared by dissolving Co(NO3)2·6H2O(3.741 g)into 150 mlofdeionized water.28%ammonia aqueous solution was added drop wise until the pH of the solution reached 11 and stirred for 30 min.3 g of ZSM-5 support was added to the resulting solution and stirred for another 1 h.All the above operations were performed at room temperature.The suspension was transferred to an oil bath preheated at 60°C,to allow for the evaporation of ammonia and decrease of pH and consequently,the deposition of cobalt species on ZSM-5.When the pH value of the suspension decreased to 6-7,the evaporation process was terminated.The solids were collected by filtration and were washed with deionized water number of times[25].All the catalysts were dried at 120°C for 12 h and calcined at 600°C for 6 h under static air in a muf fle furnace.The catalyst will be referred to as Co3O4/ZSM-5x(y),Co3O4/SBA-15(y)and Co3O4/MCM-41(y)where x denotes to SiO2/Al2O3ratio and y denotes the catalyst preparation method.
2.3.Catalyst characterization
The XRDdiffraction patterns ofallthe catalysts were obtained with a PANalytical Empyrean diffractometer using CuKαradiation and recorded in the 2θ range of 5°to 80°.Fourier-transform infrared spectra(FTIR)were recorded on a Bruker Tensor 27 spectrometer at room temperature.For the FTIR characterizations,the catalysts were dispersed in KBr to make pellets.The spectra resolution was 4 cm-1.All the spectra were recorded in the range of 4000-400 cm-1.N2adsorption-desorption isotherms of the catalysts at 77 K were measured by using Quantachrome Autosorb-1.The samples were outgassed at 523 K under vacuum for3 h before recording theirisotherms.The specific surface area of the samples was calculated according to the Brunauer-Emmett-Teller theory(BET).Total pore volumes were evaluated at relative pressures(P/Po)closed to unity.The acidity of the catalysts was detected using the temperature-programmed desorption of ammonia(NH3-TPD)using the Micrometrics Autochem II 2920 unit equipped with the thermal conductivity detector.NH3-TPD was carried out from 50 °C to 600 °C with a ramping rate of 10 °C·min-1,and the amount of desorbed NH3was monitored by a thermal conductivity detector(TCD).Surface composition ofthe catalysts was examined by X-ray photoelectron spectroscopy(XPS)analysis.XPS was performed under an ultrahigh vacuum using an ESCALAB 250Xi spectrometer with AlKαradiation(1486.6 eV)and a multichannel detector.The collected binding energies were calibrated using the C1s peak at284.6 eV as the reference.
2.4.Reaction procedure and product analysis
The thermaldecomposition reaction was carried out in a 1L stainless steel autoclave.In a typical reaction,HDC(10.0 g)and chlorobenzene(560.0 g)were charged into the autoclave containing catalyst(0.55 g).Then,the autoclave was purged with N2(99.9%)at the flow rate of 800 ml·min-1for 15 min to ensure the complete removal of inner oxygen.The reaction mixture was then heated to 230°C at the heating rate of 10 °C·min-1and held at the temperature with continuous mechanical stirring for 3 h.The reaction time started once the autoclave reached the desired temperature.During the reaction,the methanol produced by the decomposition ofHDC to HDIwas continually removed from the reaction system using N2flow at 800 ml·min-1.The pressure of the autoclave was 0.68 MPa.After the reaction,the autoclave was cooled to room temperature.The catalyst was separated by simple centrifugation from the reaction mixture and the product mixture sample was taken for analysis.The product mixture collected was quantitatively analyzed by Shimadzu GC-2010 equipped with a Rtx-5(30 m×0.25 mm×0.25 μm)capillary column and flame ionization detector(FID).The injection and detector temperatures were 260°C and 280°C,respectively.The temperature program for GC analysis was held at 150 °C for 6 min,from 150 °C to 230 °C at 10 °C·min-1and then held at 230°C for 5 min.Nitrogen and Biphenylwere used as a carrier gas and internal standard for quantitative analysis,respectively.HDC,HMI and HDI standard curves were conducted under the same conditions.The HMI standard curve was accomplished by the mass balance technique.The relative standard deviation(RSD)for the same sample through five sampling analyses by GC was less than 0.3%.HDC conversion(XHDC),HDIyield(YHDI)and HMIyield(YHMI)was calculated according to the Eqs.(1)-(3):

where CHDCis the concentration of HDC in the product sample,ppm;CHDIis the concentration of HDI in the product sample,ppm;CHMIis the concentration of HMI in the product sample,ppm;mHDCis the mass of HDC fed to the autoclave,g;mpsis the mass of product sample in the solution for quantitative analysis,g;msis the total mass of the solution for quantitative analysis,g;mpis the total mass of product,g;MHDCis the molecular weightof the HDC,g·mol-1;MHDIis the molecular weight of the HDI,g·mol-1;MHMIis the molecular weight of the HMI,g·mol-1.
3.Results and Discussion
3.1.Catalyst screening
The catalyst screening experiments for the thermal decomposition reaction was carried out under the same reaction conditions and the performances of the catalysts are compared in Table 1.At first,a blank experiment was conducted without catalyst,which gave hardly 82.1%HDC conversion and HDI yield of 28.4%with a high HMI yield of 49.5%after 3 h(Entry 1).It is worth noting that thermal decomposition of HDC could be carried out,but facilitated only to intermediate HMI rather than the desired product HDIand an efficientcatalystwas necessary to improve the HDI yield further.Therefore,different metal oxide supported ZSM-5 catalysts(M/ZSM-525(IWI),M=Co3O4,ZnO,Cr2O3)were tested.It was clearly seen that Co3O4/ZSM-525(IWI)catalyst showed a much higher HDI yield(82.3%,Entry 5)than ZnO/ZSM-525(IWI)(53.3%,Entry 3)and Cr2O3/ZSM-525(IWI)(12.5%,Entry 4)catalyst under the same conditions.Compared to ZSM-5 catalyst(Entry 2),an increase of 28.1%in HDI yield and a decrease of 27.3%in HMI yield were observed on Co3O4/ZSM-525(IWI)catalyst.This implied that the incorporation of Co3O4on ZSM-525not only induced a suitable combination of Lewis and Bronsted acid sites on the surface of the catalyst,but also boosted the conversion of HMI to HDI.Consequently,Co3O4can be considered as a suitable species over ZSM-525in the thermal decomposition of HDC.
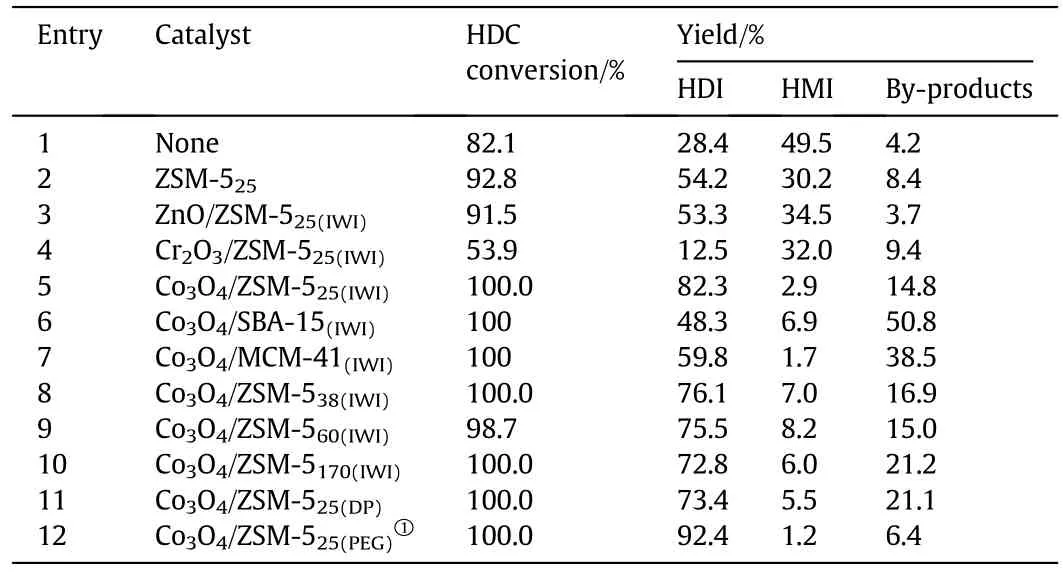
Table 1 Catalysts performance for thermal decomposition of HDC
The effect of the supports was then explored in thermal decomposition reaction using microporous and mesoporous material such as ZSM-525,SBA-15 and MCM-41 as support.Regardless ofthe type ofsupport used,HDC was completely decomposed under the same reaction conditions.Whereas,the HDI and by-products yield were significantly affected by the type of support.Compared with Co3O4/ZSM-525(IWI),significantly low HDI yield of 48.3%and 59.8%and high by-products yield of 50.8%and 38.5%were observed on Co3O4/SBA-15(IWI)and Co3O4/MCM-41(IWI)respectively(Entries 6,7).Such results suggested that microporous ZSM-525as support was more selective to HDI and less selective to by-products as compared to mesoporous SBA-15 and MCM-41 as support.Thus,the combination of Co3O4and ZSM-525was the most effective for the thermal decomposition of HDC.
Given that Co3O4/ZSM-5 catalyst facilitated desire product,so attempts were made to improve the catalytic performance of Co3O4/ZSM-5.First,Co3O4/ZSM-5 catalysts with different SiO2/Al2O3ratios were studied in the thermal decomposition of HDC.The HDI yield was increased and by-products yield was decreased with decreasing the SiO2/Al2O3ratios(Entries 5,8-10).Among the ZSM-5 supports evaluated in this study,ZSM-525was again shown to be the support with highest catalytic performance.Although Co3O4/ZSM-525(IWI)catalyst showed high performance,the HDI yield was still lower than 90%with a relatively high by-products yield of~15%.Zengzan et al.reported that catalyst preparation method for Co3O4/ZSM-5 catalyst has a strong in fluence on its physicochemical properties and caused to improve its catalytic performance[26].Given our interest in high HDI yield and low by-products yield,Co3O4/ZSM-5 catalystwas prepared by DP method and PEG method and employed in the thermal decomposition of HDC.The Co3O4/ZSM-525(DP)exhibited inferior performance than the Co3O4/ZSM-525(IWI)catalyst.Only 73.4%HDI yield with by-products yield of 21.1%was achieved on Co3O4/ZSM-525(DP)catalyst after 3 h(Entry 11).Compared to Co3O4/ZSM-525(IWI)catalyst,Co3O4/ZSM-525(PEG)catalyst displayed significantly higher catalytic performance.In particular,the HDI yield of 92.4%was observed over Co3O4/ZSM-525(PEG)catalyst with an only by-products yield of 1.2%(Entry 12).To the best of our knowledge,this is the first time that such a high HDC conversion and HDI yield were achieved by the thermal decomposition of HDC.
All these findings showed that the Co3O4/ZSM-525catalysts revealed superior catalytic performances compare to other metal oxide supported on ZSM-5 catalysts and Co3O4supported on SBA-15,MCM-41 catalysts.Therefore,Co3O4/ZSM-525catalysts were characterized to gain further insights into the in fluence of the preparation method on their surface properties and understand the possible reason for high performance.
3.2.Characterization of the catalyst
The XRD patterns of bare ZSM-525and different Co3O4/ZSM-525catalysts were performed and shown in Fig.2.All the catalysts revealed characteristic peaks of ZSM-525crystal in the ranges of 2θ=7°-9°and 2θ =23°-25°which were evident that the crystalline framework of the ZSM-525was retained after the loading in all the catalysts[27].There were no characteristic peaks of the CoO phase observed at 2θ =34.1°,39.5°,57.2°and 68.4°[26].Whereas,the diffraction peaks of the cubic spinel phase Co3O4were observed at 2θ =19.0°,31.2°,36.8°,38.5°,44.8°,59.4°and 65.2°corresponding to the(111),(220),(311),(222),(400),(511)and(440)crystal faces(JCPDS 42-1467)in the Co3O4/ZSM-525(IWI)and Co3O4/ZSM-525(PEG)catalyst.The diffraction peak intensity of the Co3O4phase was found higher in Co3O4/ZSM-525(PEG)as compared to Co3O4/ZSM-525(IWI)catalyst.These results apparently suggested that the PEG method facilitated the deposition of most Co3O4particles on the surface of ZSM-525,while the IWI method loaded some of the particles in the channels of ZSM-525.On the other hand,the diffraction peaks of Co3O4were not detected in the Co3O4/ZSM-525(DP)catalyst,it could be considered that cobalt species were well dispersed on the ZSM-5 support,or aggregated crystallites were too small to be detected by XRD.
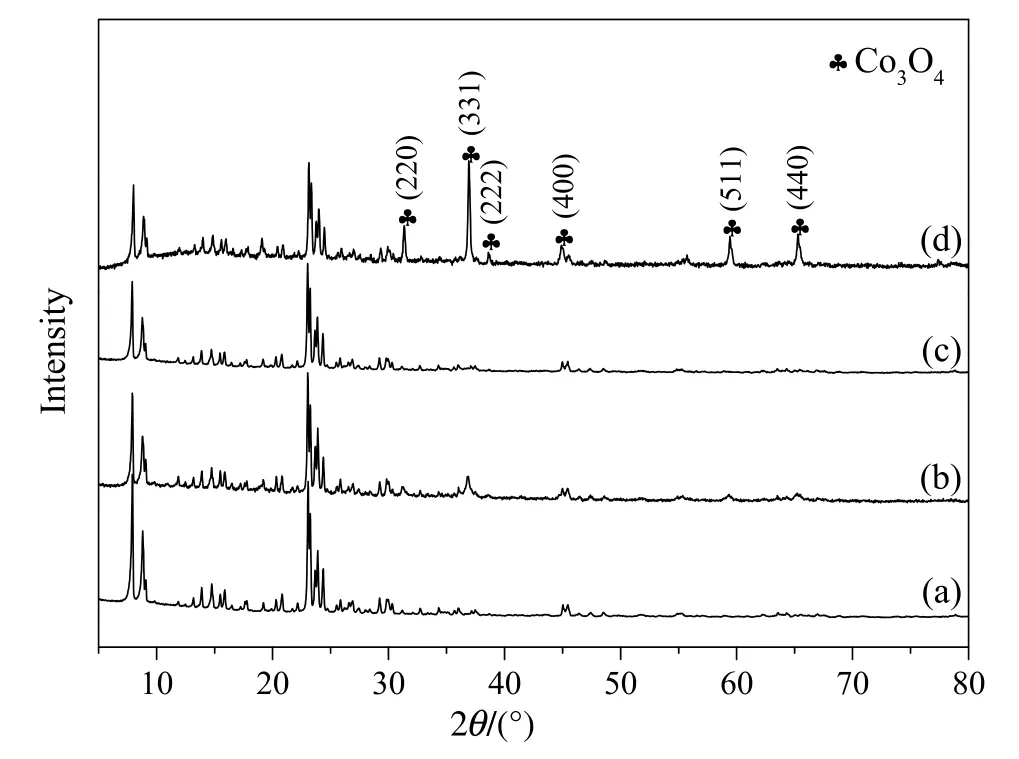
Fig.2.XRD diffraction patterns of bare ZSM-525(a),Co3O4/ZSM-525(IWI)(b),Co3O4/ZSM-525(DP)(c)and Co3O4/ZSM-525(PEG)(d)catalyst.
FTIR spectroscopic studies were carried out to investigate the in fluence of cobalt on the zeolite framework in Co3O4/ZSM-525catalysts.FTIR spectra of bare ZSM-525and Co3O4/ZSM-525catalysts are shown in Fig.3.The bare ZSM-525and Co3O4/ZSM-525catalysts exhibited band around 3644,3438 and 3220 cm-1,which were assigned to terminal hydroxyl groups Si-OH,hydroxylgroups on extra framework aluminum and strong Bronsted acid sites Si-OH-Al groups on the zeolite's surface[28].The waterbending vibration ataround 1630 cm-1wasobserved for bare ZSM-525and Co3O4/ZSM-525catalysts.This vibration was intensified in Co3O4/ZSM-525catalysts because of the presence of adsorbed water that might take place during the catalyst preparation process.The bare ZSM-525revealed the characteristic bands of MFI structure ataround 1220,1083,796,544 and 452 cm-1,which were attributed to external asymmetric stretch,internal asymmetric stretch,external symmetric stretch,double five ring,and T-O bending vibration of internal tetrahedral respectively[29-32].The Co3O4/ZSM-525catalysts exhibited also the characteristic bands of MFI structure in the FTIR spectra.This result indicated that the introduction of the Co on ZSM-525had no significant in fluence on the framework of ZSM-525.
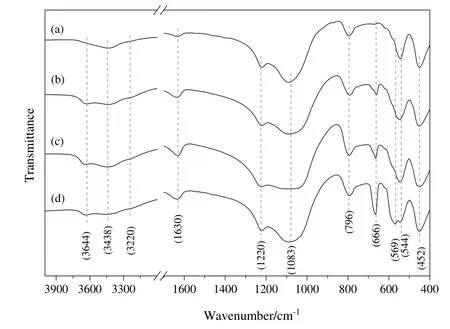
Fig.3.FTIR spectra of bare ZSM-525(a),Co3O4/ZSM-525(IWI)(b),Co3O4/ZSM-525(DP)(c)and Co3O4/ZSM-525(PEG)(d)catalyst.
Compared with the bare ZSM-525,two new bands at 569 and 666 cm-1appeared in the spectrum of Co3O4/ZSM-525catalysts.These bands can be associated with the metal-oxygen bonding which resulted after the Co loading on the ZSM-525.According to the literature[33],the band at 569 cm-1was ascribed to the Co3+octahedralcoordination,while the band at666 cm-1was ascribed to the Co2+tetrahedral coordination,confirming the formation of Co3O4cubic spinel structure.These results were in good accordance with the observation obtained from XRD study(Fig.2)that Co3O4particles with cubic spinel structure were deposited on the ZSM-525and ZSM-525structure was retained after loading cobalt on ZSM-525.
Textural properties of the bare ZSM-525and Co3O4/ZSM-525catalysts are listed in Table 2.The bare ZSM-525exhibited higher surface area(355.3 m2·g-1)and total pore volume(0.22 cm3·g-1).With the addition of cobalt oxide on ZSM-525,the resultant Co3O4/ZSM-525catalysts exhibited a decrease in surface area and total pore volume.This could be attributed to a clogging or covering of the pores of ZSM-525by Co3O4loading.The BET surface area and total pore volume of the Co3O4/ZSM-525catalysts also depended on the preparation method[26].As can be observed from Table 2,after loading cobalt oxide,the decrease in the BET surface area of Co3O4/ZSM-525catalysts was very different and varied with preparation method.The Co3O4/ZSM-525(IWI)catalyst had a lowest BET surface area(131.9 m2·g-1)and total pore volume(0.09 cm3·g-1)among other Co3O4/ZSM-525catalysts.The significant decrease in surface area and total pore volume could be as a result of pore blockage by Co3O4species.The Co/ZSM-525(DP)catalyst had less decrease in the BET surface area(241.1 m2·g-1)and total pore volume(0.16 cm3·g-1)than Co3O4/ZSM-525(IWI)catalyst.The BET surface area(254.4 m2·g-1)and total pore volume(0.18 cm3·g-1)of the Co3O4/ZSM-525(PEG)catalyst was higher than that of the Co3O4/ZSM-525(DP)catalyst.The high surface area and total pore volume of the Co3O4/ZSM-525(PEG)catalyst could be attributed to the deposition of Co species on the support surface,resulting in less Co3O4species entering into the pores.
Temperature programmed desorption of ammonia(NH3-TPD)was used to measure the acidity and acidic sites distribution of the Co3O4/ZSM-525catalysts.The amounts of acidity are listed in Table 2 and the acidic sites distribution is shown in Fig.4.NH3-TPD pro files for bare ZSM-5 and Co3O4/ZSM-525catalysts showed two major desorption zones approximately at 50 °C to 300 °C for week acidic sites and 300 °C to 600 °C for strong acidic sites.As can be seen in Fig.4,the TPD pro files were significantly varied with the catalyst preparation method.Itwas clearly observed thatthe addition ofcobaltoxide on ZSM-525by differentpreparation methods,directed to an increase of total acidity compared with bare ZSM-525(Table 2).The total acidity increased over Co3O4/ZSM-525catalysts in the order of Co3O4/ZSM-525(DP)<Co3O4/ZSM-525(IWI)<Co3O4/ZSM-525(PEG)which was well consistence with the HDI yield(Table 1).It meant that the acidity of Co3O4/ZSM-525played a significant role in the catalytic performance and the Co3O4/ZSM-525(PEG)catalyst with high total acidity revealed high HDI yield.From the aspects of acidic sites distribution,all the Co3O4/ZSM-525catalysts except for Co3O4/ZSM-525(PEG)catalyst clearly exhibited higher weak acidic sites than strong acidic sites(Table 2).In particular,Co3O4/ZSM-525(DP)catalyst revealed an extra board NH3desorption peak centered at 530°C,which might be attributed to the formation of very strong acidic sites.Whereas,the Co3O4/ZSM-525(PEG)catalyst showed almost equal weak acidic sites and strong acidic sites on the surface,which might be another reason for the superior catalytic performance.
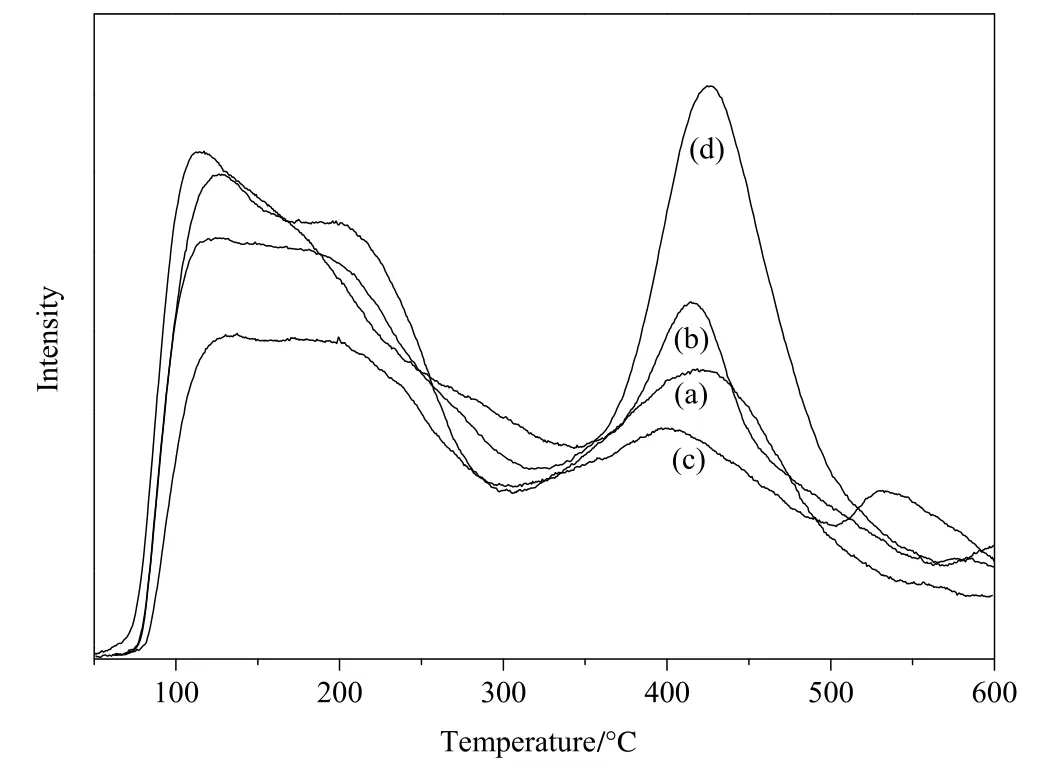
Fig.4.NH3-TPD pro files of bare ZSM-525(a),Co3O4/ZSM-525(IWI)(b),Co3O4/ZSM-525(DP)(c)and Co3O4/ZSM-525(PEG)(d)catalyst.
The Co3O4/ZSM-525catalysts were also characterized by XPS to investigate the in fluence of the preparation method on their surface properties.The XPS spectra of Co2p and O1s are shown in Fig.5,and the XPS analysis data are summarized in Table 3.The Co3O4/ZSM-525catalysts had two main peaks at 780.5-780.7 eV and 795.4-795.7 eV in their XPS Co2p spectra,which were attributed to Co2p3/2and Co2p1/2spin-orbital peaks respectively(Fig.5A).It was hard to distinguish Co2+and Co3+from the Co2p spectra due to the small difference in their binding energy value.Whereas,the spin-orbit splitting value of Co2p was found to well correlate with the Co oxidation state.It was reported that the spin-orbits plitting value for Co3+compounds is 15.0 eV[34].For the mixed-valence Co3O4,spin-orbit splitting value 15.1-15.3 eV has been reported[35].Here,the spin-orbit value was about 15.1 eV for Co3O4/ZSM-525catalysts(Table 3),which was close to that of mixed-valence Co3O4and suggested that the cobalt species were present mainly as Co3O4,on the cobalt oxide supported on ZSM-525catalysts.
As can see in Table 3,the catalyst preparation method had a clear effect on the surface composition.The results showed that the surface cobalt content of the Co/ZSM-525catalyst increased in the order of Co3O4/ZSM-525(DP)<Co3O4/ZSM-525(IWI)<Co3O4/ZSM-525(PEG).The surface cobalt content on the Co3O4/ZSM-525(PEG)was 16.3%,which ascribed to the cobalt species enrich menton the surface by PEG method as compared to other catalyst preparation methods.Moreover,the Co2p3/2could be deconvoluted to the two components at 779.7 and 781.9 eV,corresponding to the Co3+and Co2+respectively.The Co2p1/2could be also deconvoluted to two components at 794.8 and 796.9 eV,which could be ascribed to the Co3+and Co2+respectively.The surface content ratios of Co3+/Co2+and the relative surface content of Co3+(Co3+/(Co3++Co2+))are also listed in Table 3.As can see from the Table 3,the preparation method had an obvious influence on the surface contentratio of Co3+/Co2+and the relative surface content of Co3+.Thesurface content ratio of Co3+/Co2+(1.4%)and the relative surface content of Co3+(58.3%)of the Co3O4/ZSM-525(PEG)catalyst suggested that the surface content of Co3+was higher than other Co3O4/ZSM-525catalysts and changed in the order of Co3O4/ZSM-525(PEG)>Co3O4/ZSM-525(IWI)>Co3O4/ZSM-525(DP).

Table 2 Physicochemical properties of ZSM-525 and Co3O4/ZSM-525 catalysts
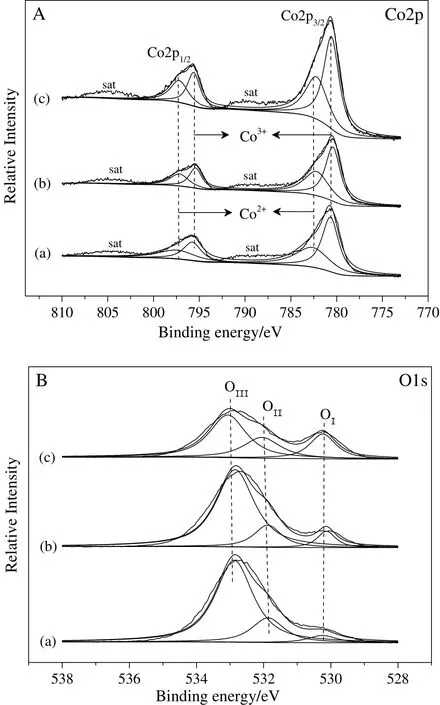
Fig.5.XPS spectra of Co2p(A)and O1s(B)of Co3O4/ZSM-525(DP)(a),Co3O4/ZSM-525(IWI)(b)and Co3O4/ZSM-525(PEG)(c)catalyst.
The Si/Al and Co/Al ratios of the Co3O4/ZSM-525catalysts from the XPS data are shown in Table 3.The Si/Al ratio on the surface of the Co3O4/ZSM-525catalysts decreased in the order of Co3O4/ZSM-525(DP)>Co3O4/ZSM-525(IWI)>Co3O4/ZSM-525(PEG),indicated enrichment of the surface with Al.The Al surface enrichment could be due to the formation of Co silicates outside the ZSM-525,thereby decreasing the Siconcentration at the zeolite surface[36].The Co/Alratio on the surface of the Co3O4/ZSM-525catalysts varied in the order of Co3O4/ZSM-525(PEG)>Co3O4/ZSM-525(IWI)>Co3O4/ZSM-525(DP),demonstrating the preferential cobalt deposition on the surface in the Co3O4/ZSM-525(PEG)catalyst,as discussed earlier in this section.
The XPS spectra of O1s for Co3O4/ZSM-525catalysts are also presented in Fig.5B,could be deconvoluted to three components.The firstcomponent at 529.9 eV corresponded to the surface lattice oxygen of the Co3O4/ZSM-525catalysts(OI),the second component at 531.1 eV corresponded to the adsorbed oxygen of the Co3O4/ZSM-525catalysts(OII)and the last componentat 532.7 eV corresponded to the lattice oxygen of the ZSM-525support(OIII)[26,37].The content of OIand OIIon the surface of Co3O4/ZSM-5 catalysts varied with the preparation method as shown in Table 3.The Co/ZSM-525(PEG)catalyst had almost equal surface OIcontent(12.2%)and OIIcontent(14.5%),but higher than other catalysts.The oxygen surface composition of the solid always played a significant role in the catalytic performance.The surface lattice oxygen content for Co3O4/ZSM-525increased in the order of Co3O4/ZSM-525(DP)<Co3O4/ZSM-525(IWI)<Co3O4/ZSM-525(PEG).
3.3.Physicochemical properties-performance correlation
For cobalt oxide supported on ZSM-525catalysts,a number of parameters of catalyst such as the surface area,the surface cobalt content,the oxygen species present on the surface and the total acidity of the surface,could affect the catalytic performance[26].Therefore,the physicochemical properties of the Co3O4/ZSM-525catalysts were correlated with the HDI selectivity to gain further insights into the possible reason for high performance.The Co3O4/ZSM-525(PEG)catalyst with high surface area exhibited a high selectivity to HDI,but the Co3O4/ZSM-525(IWI)catalyst with a low surface area revealed a high selectivity to HDI compare to Co3O4/ZSM-525(DP)catalyst(Table 2).Thus,the HDI selectivity could not be correlated to the surface area of the Co3O4/ZSM-525catalyst.Herein,the correlation between the surface lattice oxygen content and the HDI selectivity could be found and followed the order of Co3O4/ZSM-525(PEG)>Co3O4/ZSM-525(IWI)>Co3O4/ZSM-525(DP)(Tables 2,3).The catalyst with more surface lattice oxygen content revealed a high HDI selectivity.It meant that the concentration of surface lattice oxygen content was a significant factor to sustain the higher catalytic performance of Co3O4/ZSM-525catalyst.
In addition,the selectivity of Co3O4/ZSM-525catalysts to HDI could also be identified by anotherrelationship,involving NH3-TPDand XPS results.The total acidity and the relative surface content of Co3+(Co3+/(Co3++Co2+))were linearly related to the HDI selectivity over Co3O4/ZSM-525catalysts as shown in Fig.6.The relationship was shown that the total acidity and the relative surface content of Co3+on Co3O4/ZSM-525catalyst were also important factors for achieving high HDI selectivity.The HDI selectivity over Co3O4/ZSM-525catalysts varied in the order of Co3O4/ZSM-525(PEG)>Co3O4/ZSM-525(IWI)>Co3O4/ZSM-525(DP).On the basis of the fact mentioned above,it could be concluded that theCo3O4/ZSM-525(PEG)catalyst had high total acidity(Table 2),high surface content of Co3+and surface lattice oxygen content(Table 3),which were responsible factors for the high HDI selectivity(92.4%).

Table 3 XPS data of Co3O4/ZSM-525 catalysts
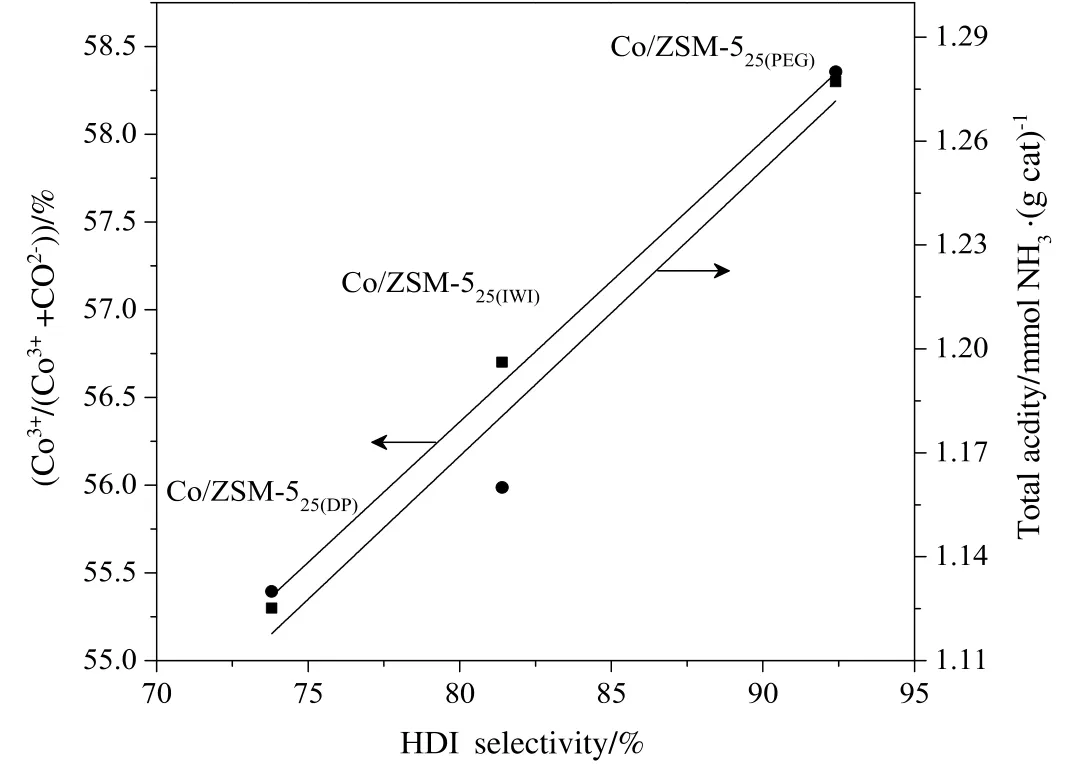
Fig.6.The relation of HDIselectivity to the relative surface contentofCo3+(Co3+/(Co3++Co2+))and total acidity on the Co3O4/ZSM-525 Catalysts.
3.4.Catalyst reusability
The ease of separation,deactivation and reusability of the heterogeneous catalyst has been always a crucial issue that must be taken into consideration during the utilization of catalyst.Consequently,recycling experiments were carried out to investigate the stability of the Co3O4/ZSM-525(PEG)catalyst under the conditions mentioned in Fig.7.In each cycle,the used catalyst was separated by simple centrifugation and washed with abundant amounts of chlorobenzene and toluene to get rid of the products adhering to the surface of the catalyst,dried at 120°C for 12 h then reused directly for the next run without regeneration.Fig.7 revealed the performance of the reused Co3O4/ZSM-525(PEG)catalyst.It was found that the Co3O4/ZSM-525(PEG)catalyst could be recovered and reused in further reactions without degradation in the performance.The HDC conversion and HDI yield remained almost unchanged over five successive runs.

Fig.7.Reusability of Co3O4/ZSM-525(PEG)catalyst in the thermal decomposition of HDC to HDI.Reaction condition:HDC concentration in chlorobenzene,1.8%;catalyst,0.55 g;temperature 230 °C;nitrogen flow rate,800 ml·min-1;pressure,0.68 MPa;time 2.5 h.
In order to investigate the preservation of Co3O4species and structure of ZSM-5,XRD was performed for fresh and used(Run 1 and Run 5)Co3O4/ZSM-525(PEG)catalyst as shown in Fig.8.The characteristic diffraction peaks in the ranges of 2θ=7°-9°and 2θ =23°-25°of used Co3O4/ZSM-525(PEG)catalyst were the same as those of fresh Co3O4/ZSM-525(PEG)catalyst.However,the intensities of the peaks of used Co3O4/ZSM-525(PEG)catalyst were weaker than those for fresh Co3O4/ZSM-525(PEG)catalyst.This indicated that the used Co3O4/ZSM-525(PEG)catalyst retained the MFI type structure,but its crystallinity slightly decreased.In addition,the diffraction peaks of the cubic spinel phase Co3O4were observed in the used catalyst,same as that of the fresh Co3O4/ZSM-525(PEG)catalyst.Thus,it could be concluded that the Co3O4species and ZSM-525structure in Co3O4/ZSM-525(PEG)catalyst,were preserved and remained active for the reaction after five successive runs.

Fig.8.XRD diffraction patterns of fresh Co3O4/ZSM-525(PEG)(a),used Run 1 Co3O4/ZSM-525(PEG)(b),used Run 5 Co3O4/ZSM-525(PEG)(c)catalyst.
3.5.Effect of reaction parameters
The Co3O4/ZSM-525(PEG)catalyst exhibited superior performance compared to other studied catalysts.Therefore,it was selected as a system catalyst for the evaluation of the thermal decomposition reaction parameters to obtain best reaction conditions for the high yield of HDI.Various parameters such as reaction temperature,HDC concentration in solvent,catalyst content and reaction time were studied.
3.5.1.Effect of reaction temperature
The effect of reaction temperature on the thermal decomposition of HDC was studied and the results are shown in Fig.9.The reaction temperature was varied from 200 °C to 250 °C.It was observed that the reaction temperature exhibited a dramatic effect on the reaction yield.The HDC conversion and HDI yield were found to increase with increasing reaction temperature.Particularly,HDC conversion and HDI yield at 200°C dropped to 89.3%and 43.4%,respectively,whereas the Co3O4/ZSM-525(PEG)catalyst showed maximum activity at a reaction temperature of 250°C after 2 h.HDC conversion reached 100%with an HDI yield of 98.4%in the presence of Co3O4/ZSM-525(PEG)catalyst at 250°C after 2 h.The reaction temperature higher than 250°C was not feasible because HMI almost completely decomposed to HDI and possibly led to formation of by-products involving the addition and condensation polymerization of HDI[14].Regarding the by-products formation and energy concerns,the reaction temperature higher than 250°C should be avoided to achieve maximum HDI yield.Hence,250°C was selected as suitable reaction temperature.
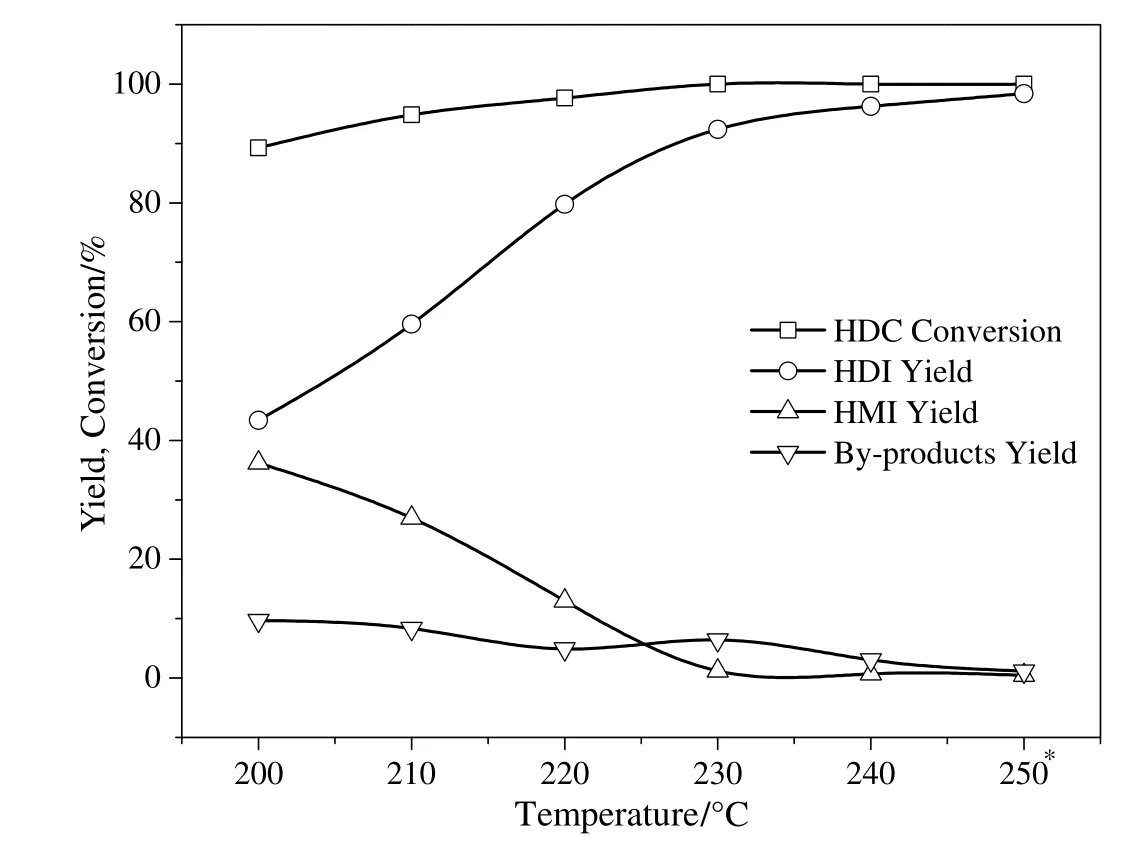
Fig.9.Effect of reaction temperature on the thermal decomposition of HDC to HDI.Reaction condition:HDC concentration in chlorobenzene,1.8%;catalyst,0.55 g;nitrogen flow rate,800 ml·min-1;time 2.5 h.*Reaction time 2 h.
3.5.2.Effect of HDC concentration
With such results in hand,we then decided to investigate the effect of HDC concentration on the thermal decomposition of HDC over the Co3O4/ZSM-525(PEG)catalyst.The decomposition reaction was performed by varying the HDC concentration(1.8%-10%).As inferred from Fig.10,with the increase in HDC concentration,the HDC conversion and HDI yield remain almost same in the range of 1.8%-6%and then decrease gradually in the range of 6%-10%.The HDC conversion was 100%with an HDI yield of 95.7%and HMI yield of 2.1%,when 6.0%HDC concentration was used.It can be clearly observed that when the HDC concentration increased to 6.5%,the HDC conversion and HDI yield decreased to 98.2%and 85.6%respectively while HMI yield increased to 12.1%.
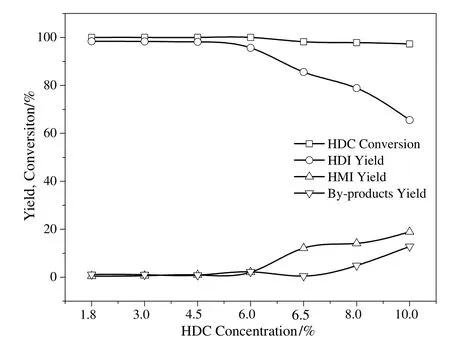
Fig.10.Effect of HDC conversion on the thermal decomposition of HDC to HDI.Reaction condition:catalyst,0.55 g;temperature 250 °C;nitrogen flow rate,800 ml·min-1;pressure,1.0 MPa;time 2 h.
The HDC conversion and HDI yield continued to decrease with the increase in HDC concentration and reached 97.3%and 65.6%respectively,at 10%HDC concentration.Comparatively,an opposite trend in the HMI and by-products yield was observed as shown in Fig.10.The HMI and by-products yield increased consistent with the increased HDC concentration.As reported in the literature,this phenomenon took place at high HDC concentrations probably due to the large extent of disproportion of HDC and carbonization of HDI[11,13].From economic and product yield viewpoint,a higher HDC concentration was not preferable to the HDI production and a too low HDC concentration usually meant a lower efficiency of the production process.Therefore,6.5%HDC concentration was considered to be the optimum concentration for HDI synthesis.
3.5.3.Effect of catalyst content
The study was then extended to investigate the effectofcatalystcontenton the thermal decomposition of HDC over the Co3O4/ZSM-525(PEG)catalyst.The decomposition reaction was accomplished by using different catalyst content(0.5 wt%-5 wt%of HDC).It can be seen from Fig.11,the HDC conversions were all 100%,regardless of the catalyst content ranging from 0.5 wt%to 5 wt%.
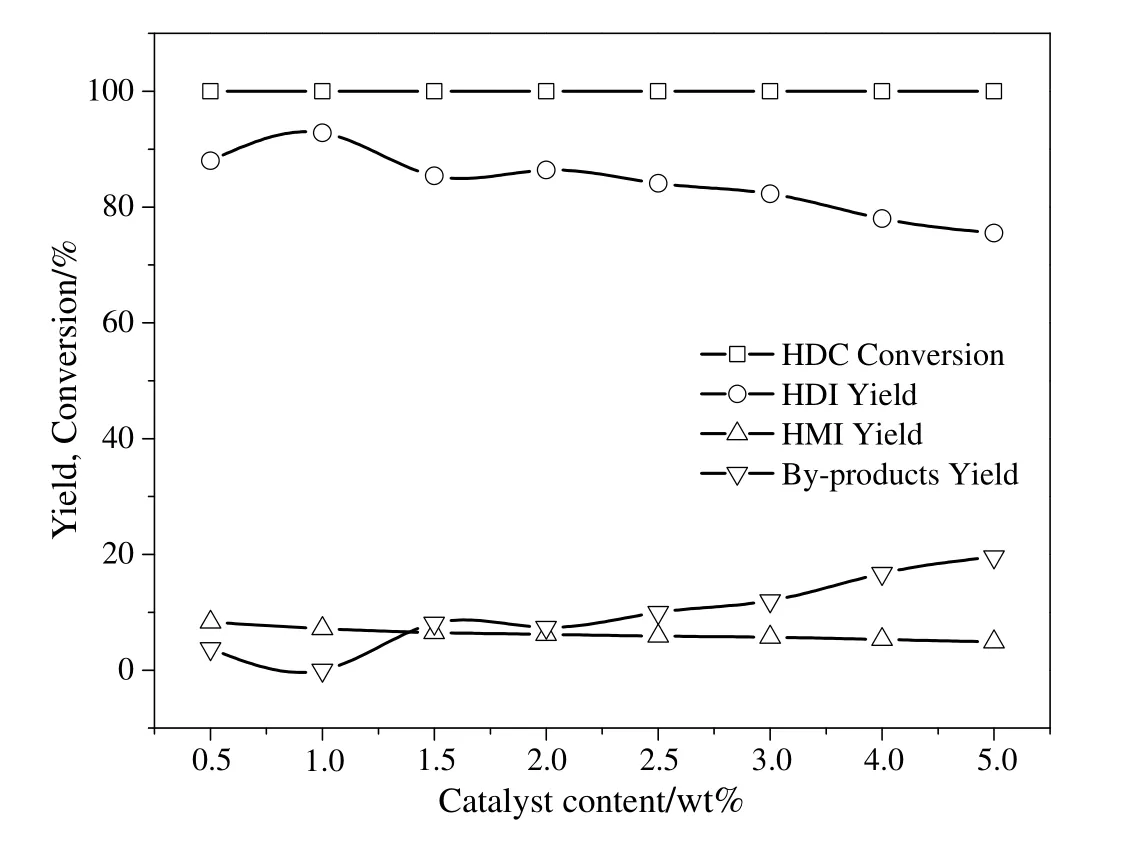
Fig.11.Effect of catalyst content on the thermal decomposition of HDC to HDI.Reaction condition:HDC concentration in chlorobenzene,6.5%;temperature 250°C;nitrogen flow rate,800 ml·min-1;pressure,1.0 MPa;time 2.5 h.
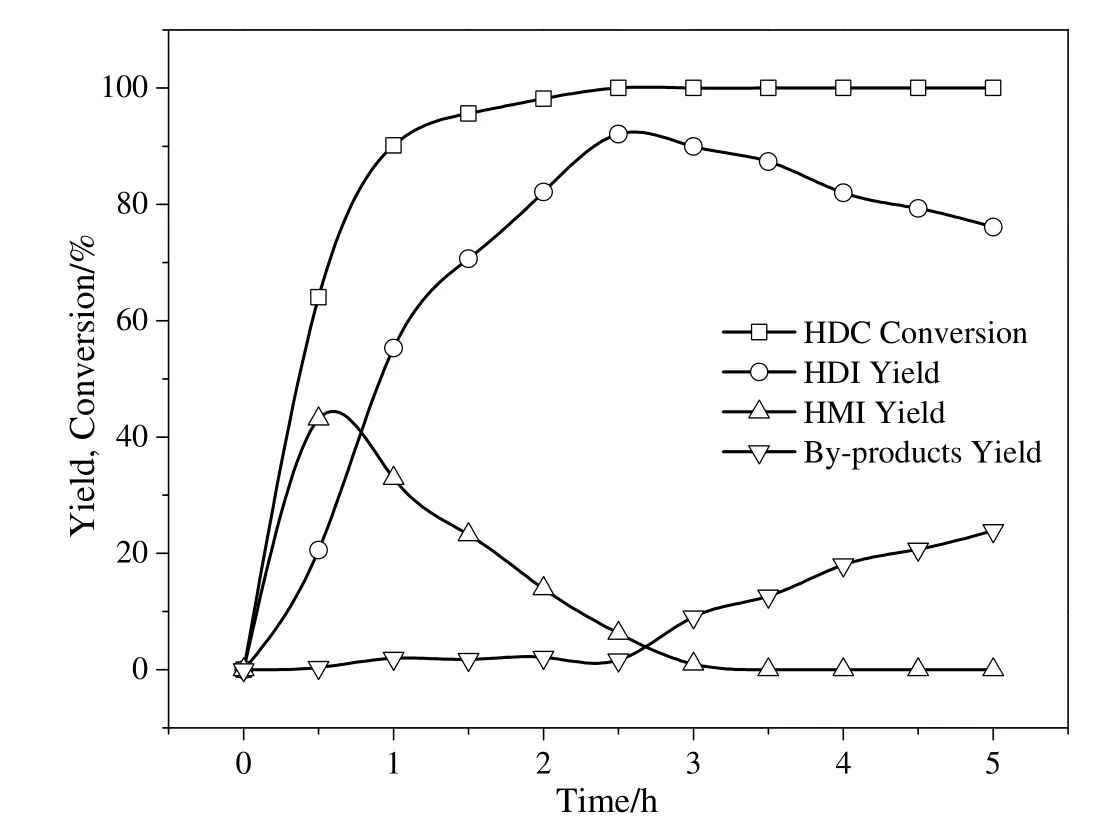
Fig.12.Effect of reaction time on the thermal decomposition of HDC to HDI.Reaction condition:HDC concentration in chlorobenzene,6.5%;temperature 250°C;catalyst,1 wt%of HDC concentration;nitrogen flow rate,800 ml·min-1;pressure,1.0 MPa.
The most effective thermal decomposition of HDC was observed with the 1 wt%catalyst content which revealed HDI yield of 92.8%and HMI yield of 7.2%.The HDI yield decreased with the increase in catalyst content from 1 wt%to 5 wt%and reached 75.5%at 5 wt%catalyst content.However,the HMI yield decreased only marginally from 1 wt%to 5 wt%catalyst content.While,the by-products yield was found to increase with the increase in catalyst content from 1 wt%to 5 wt%and 19.6%was observed at 5 wt%catalyst content.These observations indicated that higher catalyst content in the system provided an extra stoichiometric amount of protons leading to HDI polymerization and high by-products yield.On the other hand,the HDI yield decreased to 88.0%and HMIand by-products yield increased to 8.3%and 3.7%respectively in the presence of 0.5 wt%catalyst content.The decrease in the HDI yield and increase in the HMI yield might be due to reduced accessibility to active sites.In this work,1 wt%catalyst content was observed to be preferable for HDC thermal decomposition reaction.
3.5.4.Effect of reaction time
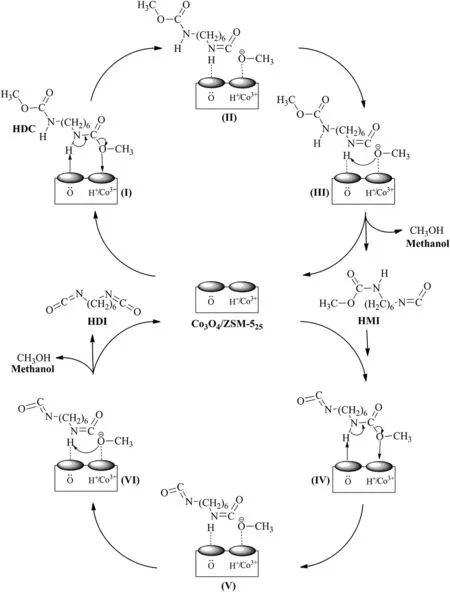
Fig.13.Possible reaction mechanism for the thermal decomposition of HDC to HDI over Co3O4/ZSM-525 catalyst.
The effect of reaction time on the thermal decomposition of HDC over the Co3O4/ZSM-525(PEG)catalyst was investigated and results are shown in Fig.12.Initially,HDC conversion sharply increased with increase in reaction time and reached 90%after 1 h.The HDI and HMI yield were 55.3%and 32.9%respectively after 1 h.However,when the reaction proceeded for 2.5 h,the HDC conversion increased to 100%and then maintained after 5 h.The HDI yield gradually increased and HMI yield decreased with the reaction time increase from 1 h to 2.5 h,and reached to>92%and 6.3%,respectively.With prolonged reaction time,nearly no HMI was observed,which meant that HMI was completely decomposed to HDI.However,the HDI yield dropped and by-products yield increased due to side reactions when the reaction time increased from 2.5 h to 5 h.It indicated that longer reaction time was not favorable to HDI synthesis.Thus,the optimum reaction time for thermal decomposition of HDC was 2.5 h.
3.6.Possible reaction mechanism
As described earlier,the performance ofthe Co3O4/ZSM-525catalysts could be related to surface content of Co3+,surface lattice oxygen content and total acidity which provided greater availability of the active phase for the reaction.Therefore,based on the physicochemical properties of Co3O4/ZSM-525catalysts,the thermal decomposition of HDC to HDI possibly took place over Co3O4/ZSM-525as shown in Fig.13.The thermal decomposition of HDC to HDI over Co3O4/ZSM-525involved two stages(Fig.1),HDC first converted to HMI and then conversion of HMI to HDI.Both stages followed the same mechanism steps.
First,HDC molecule was activated through the adsorption of hydrogen atom in amide group onto the surface lattice oxygen and oxygen atom in methoxy group onto the surface acidic site of the catalyst(species I).The hydrogen atom in amide group and oxygen atom in methoxy group were cleaved and attached to surface lattice oxygen and surface acidic site respectively by coordinate bonds in the species II.Meanwhile,intermediate HMI was generated.Consequently,the hydrogen atom at the surface lattice oxygen via coordinate bond was attacked by the oxygen atom in methoxy group at the surface acidic site(species III),resulting in rupture of coordinate bonds to form CH3OH and the Co3O4/ZSM-525catalyst was regenerated.The carbamate group of HMI molecule was further attacked by Co3O4/ZSM-525catalyst again,and similarly final product HDI was synthesized by the above mechanism steps.
4.Conclusions
In conclusion,a green route for the synthesis of HDI by thermal decomposition of HDC was successfully developed over cobalt oxide supported ZSM-525catalyst,using low boiling point solvent.The as-prepared metal oxide supported catalysts were employed as heterogeneous catalyst for thermal decomposition of HDC to HDI.Co3O4/ZSM-525catalyst outperformed the other investigated catalysts in terms ofconversion and yield.The preparation method was observed to seriously in fluence the catalytic performance of the Co3O4/ZSM-525catalysts.Following the order of Co3O4/ZSM-525(PEG)>Co3O4/ZSM-525(IWI)>Co3O4/ZSM-525(DP),the Co3O4/ZSM-525(PEG)catalyst revealed superior catalytic performance.The physicochemical properties of Co3O4/ZSM-525catalysts were characterized by various techniques and it was found that the introduction of the cobalt oxide on ZSM-525had no significant influence on the framework of ZSM-525.However,PEG method deposited the most Co3O4particles on the surface of ZSM-525as compared to other preparation method and had evident effects on the physicochemical properties of the catalyst.The superior performance of the Co3O4/ZSM-525(PEG)catalyst was attributed to higher values of surface cobalt content,relative surface content of Co3+,surface lattice oxygen content and total acidity.Under optimum reaction conditions:6.5%HDC concentration,1 wt%Co3O4/ZSM-525(PEG)catalyst,250 °C temperature,2.5 h time,800 ml·min-1nitrogen flow rate and 1.0 MPa pressure,the HDC conversion and HDI yield were reached 100%and 92.8%respectively.The Co3O4/ZSM-5PEGcatalyst showed high stability and could be recycled for five successive runs without loss of performance.
[1]D.Randall,S.Lee,The Huntsman Polyurethanes Book,Wiley,United Kingdom,2002.
[2]W.D.Vilar,Chemistry and Technology of Polyurethanes,Third updated ed.Vilar Consultoria Técnica Ltda.,Rio de Janeiro,Brazil,2002.
[3]J.S.Nowick,D.L.Holmes,G.Noronha,E.M.Smith,T.M.Nguyen,S.L.Huang,Synthesis of peptide isocyanates and isothiocyanates,J.Org.Chem.61(11)(1996)3929-3934.
[4]E.Delebecq,J.P.Pascault,B.Boutevin,F.Ganachaud,On the versatility of urethane/urea bonds:Reversibility,blocked isocyanate,and non-isocyanate polyurethane,Chem.Rev.113(1)(2012)80-118.
[5]P.Dubé,N.F.F.Nathel,M.Vetelino,M.Couturier,C.L.Aboussafy,S.Pichette,M.L.Jorgensen,M.Hardink,Carbonyldiimidazole-mediated Lossen rearrangement,Org.Lett.11(24)(2009)5622-5625.
[6]V.V.Sureshbabu,B.S.Patil,R.Venkataramanarao,Preparation,isolation,and characterization of N α-Fmoc-peptide isocyanates:Solution synthesis of oligo-α-peptidyl ureas,J.Org.Chem.71(20)(2006)7697-7705.
[7]B.Akhlaghinia,A new and convenient method of generating alkyl isocyanates from alcohols,thiols and trimethylsilyl ethers using triphenylphosphine/2,3-dichloro-5,6-dicyanobenzoquinone/Bu4NOCN,Synthesis(12)(2005)1955-1958.
[8]W.H.Lin,Y.S.Guo,S.A.Dai,An efficient one-pot synthesis of aliphatic diisocyanate from diamine and aiphenyl carbonate,J.Taiwan Inst.Chem.Eng.50(2015)322-327.
[9]G.Zhu,H.Li,Y.Cao,H.Liu,X.Li,J.Chen,Q.Tang,Kinetic study on the novel efficient clean decomposition of methyl n-phenyl carbamate to phenyl isocyanate,Ind.Eng.Chem.Res.52(12)(2013)4450-4454.
[10]Y.Cao,H.Li,N.Qin,G.Zhu,Kinetics of the decomposition of dimethylhexane-1,6-dicarbamate to 1,6-hexamethylene diisocyanate,Chin.J.Chem.Eng.23(5)(2015)775-779.
[11]T.Masuda,D.Saylik,L.Diebele,Production of Aliphatic Isocyanate,Japan Pat.,6239826,1994.
[12]J.Wildschut,F.H.Mahfud,R.H.Venderbosch,H.J.Heeres,Hydrotreatment of fast pyrolysis oil using heterogeneous noble-metal catalysts,Ind.Eng.Chem.Res.48(23)(2009)10324-10334.
[13]S.C.Miranda,C.C.Cabrero,E.F.Gutierrez,P.S.Carnero,M.S.Queralt,P.U.Sola,Isocyanate Production Procedure,US Pat.,6639101,2003.
[14]D.L.Sun,J.Y.Luo,R.Y.Wen,J.R.Deng,Z.S.Chao,Phosgene-free synthesis of hexamethylene-1,6-diisocyanate by the catalytic decomposition of dimethylhexane-1,6-dicarbamate over zinc-incorporated berlinite(ZnAlPO4),J.Hazard.Mater.266(2014)167-173.
[15]M.J.Hyun,M.Shin,Y.J.Kim,Y.W.Suh,Phosgene-free decomposition of dimethylhexane-1,6-dicarbamate over ZnO,Res.Chem.Intermed.(2015)1-14.
[16]A.Y.Khodakov,W.Chu,P.Fongarland,Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels,Chem.Rev.107(5)(2007)1692-1744.
[17]Y.Li,S.Liu,S.Xie,L.Xu,Promoted metal utilization capacity of alkali-treated zeolite:Preparation of Zn/ZSM-5 and its application in 1-hexene aromatization,Appl.Catal.A Gen.360(1)(2009)8-16.
[18]Á.Szegedi,M.Popova,C.Minchev,Catalytic activity of Co/MCM-41 and Co/SBA-15 materials in toluene oxidation,J.Mater.Sci.44(24)(2009)6710-6716.
[19]T.Kanazawa,MFI zeolite as a support for automotive catalysts with reduced Pt sintering,Appl.Catal.B Environ.65(3)(2006)185-190.
[20]L.B.Pierella,C.Saux,S.C.Caglieri,H.R.Bertorello,P.G.Bercoff,Catalytic activity and magnetic properties of Co-ZSM-5 zeolites prepared by different methods,Appl.Catal.A Gen.347(1)(2008)55-61.
[21]S.H.Kang,J.H.Ryu,J.H.Kim,P.S.Prasad,J.W.Bae,J.Y.Cheon,K.W.Jun,ZSM-5 supported cobalt catalyst for the direct production of gasoline range hydrocarbons by Fischer-Tropsch synthesis,Catal.Lett.141(10)(2011)1464-1471.
[22]A.Gervasini,Characterization of the textural properties of metal loaded ZSM-5 zeolites,Appl.Catal.A Gen.180(1)(1999)71-82.
[23]Z.M.El-Bahy,M.M.Mohamed,F.I.Zidan,M.S.Thabet,Photo-degradation of acid green dye over Co-ZSM-5 catalysts prepared by incipient wetness impregnation technique,J.Hazard.Mater.153(1)(2008)364-371.
[24]M.Yao,N.Yao,Y.Shao,Q.Han,C.Ma,C.Yuan,C.Li,X.Li,New insight into the activity of ZSM-5 supported Co and CoRu bifunctional Fischer-Tropsch synthesis catalyst,Chem.Eng.J.239(2014)408-415.
[25]L.F.Chen,P.J.Guo,M.H.Qiao,S.R.Yan,H.X.Li,W.Shen,H.L.Xu,K.N.Fan,Cu/SiO2catalysts prepared by the ammonia-evaporation method:Texture,structure,and catalytic performance in hydrogenation of dimethyl oxalate to ethylene glycol,J.Catal.257(1)(2008)172-180.
[26]Z.Zhu,G.Lu,Z.Zhang,Y.Guo,Y.Guo,Y.Wang,Highly active and stable Co3O4/ZSM-5 catalyst for propane oxidation:Effect of the preparation method,ACS Catal.3(6)(2013)1154-1164.
[27]X.Huang,B.Hou,J.Wang,D.Li,L.Jia,J.Chen,Y.Sun,CoZr/H-ZSM-5 hybrid catalysts for synthesis of gasoline-range isoparaffins from syngas,Appl.Catal.A Gen.408(1)(2011)38-46.
[28]L.F.Isernia,FTIR study of the relation,between extra-framework aluminum species and the adsorbed molecular water,and its effect on the acidity in ZSM-5 steamed zeolite,Mater.Res.16(4)(2013)792-802.
[29]X.Zhao,L.Wei,J.Julson,Q.Qiao,A.Dubey,G.Anderson,Catalytic cracking of nonedible sun flower oil over ZSM-5 for hydrocarbon bio-jet fuel,New Biotechnol.32(2)(2015)300-312.
[30]X.Zhao,L.Wei,J.Julson,Z.Gu,Y.Cao,Catalytic cracking of inedible camelina oils to hydrocarbon fuels over bifunctional Zn/ZSM-5 catalysts,Korean J.Chem.Eng.32(8)(2015)1528-1541.
[31]S.H.Zhang,Z.X.Gao,S.J.Qing,S.Y.Liu,Y.Qiao,Effect of zinc introduction on catalytic performance of ZSM-5 in conversion of methanol to light ole fins,Chem.Pap.68(9)(2014)1187-1193.
[32]N.A.S.Amin,D.D.Anggoro,Characterization and activity of Cr,Cu and Ga modified ZSM-5 for direct conversion of methane to liquid hydrocarbons,J.Nat.Gas Chem.12(2)(2003)123-134.
[33]C.W.Tang,C.B.Wang,S.H.Chien,Characterization of cobalt oxides studied by FT-IR,Raman,TPR and TG-MS,Thermochim.Acta 473(1)(2008)68-73.
[34]J.Cheng,J.Yu,X.Wang,L.Li,J.Li,Z.Hao,Novel CH4combustion catalysts derived from Cu-Co/X-Al(X=Fe,Mn,La,Ce)hydrotalcite-like compounds,Energy Fuel 22(4)(2008)2131-2137.
[35]J.Y.Luo,M.Meng,X.Li,X.G.Li,Y.Q.Zha,T.D.Hu,Y.N.Xie,J.Zhang,Mesoporous Co3O4-CeO2and Pd/Co3O4-CeO2catalysts:Synthesis,characterization and mechanistic study of their catalytic properties for low-temperature CO oxidation,J.Catal.254(2)(2008)310-324.
[36]C.Chupin,A.Van Veen,M.Konduru,J.Després,C.Mirodatos,Identity and location of active species for NO reduction by CH4over Co-ZSM-5,J.Catal.241(1)(2006)103-114.
[37]Q.Liu,L.C.Wang,M.Chen,Y.Cao,H.Y.He,K.N.Fan,Dry citrate-precursor synthesized nanocrystalline cobalt oxide as highly active catalyst for total oxidation of propane,J.Catal.263(1)(2009)104-113.
 Chinese Journal of Chemical Engineering2017年12期
Chinese Journal of Chemical Engineering2017年12期
- Chinese Journal of Chemical Engineering的其它文章
- Transport properties of dilute ammonia-noble gas mixtures from new intermolecular potential energy functions
- Solid-liquid equilibrium of dimethyl terephthalate(DMT),dimethyl isophthalate(DMI)and dimethyl phthalate(DMP)in melt crystallization process
- Analogy between adsorption and sorption:An elementary mechanistic approach.I.Monolayer adsorption and sorption without solvent cluster formation
- Permeation properties of CO2 and CH4 in asymmetric polyethersulfone/polyesterurethane and polyethersulfone/polyetherurethane blend membranes
- Pt-H2SO4/Zr-mont morillonite:An efficient catalyst for the polymerization of octamethylcy-clotetrasiloxane,poly methylhydrosiloxane and hexamethyldisiloxane to low-hydro silicone oil☆
- Research progress in the SO2 resistance of the catalysts for selective catalytic reduction of NO x☆