
Zn/SiO2气相催化裂解1,1,2-三氯乙烷脱HCl:酸性与失活
2017-05-11 00:45:57胡益浩,宋通洋,王月娟等
物理化学学报 2017年5期
Zn/SiO2气相催化裂解1,1,2-三氯乙烷脱HCl:酸性与失活
胡益浩 宋通洋 王月娟 胡庚申 谢冠群*罗孟飞*
(浙江师范大学物理化学研究所,先进催化材料教育部重点实验室,浙江金华321004)
用浸渍法制备了一系列SiO2负载的过渡金属催化剂M/SiO2(M为第Ⅳ周期过渡金属),用于气相催化裂解1,1,2-三氯乙烷(TCE)脱HCl的反应。研究发现,在M/SiO2催化剂中,Zn/SiO2催化性能最好,TCE转化率能达到98%,顺-1,2-二氯乙烯(cis-DCE)的选择性为82%。随着Zn负载量的增加,Zn/SiO2催化剂上TCE转化率逐渐增加,与催化剂上总酸量变化一致。将总酸量以Zn负载量归一化得到比酸量,则比酸量越大,Zn/ SiO2催化剂比活性越高,表明Zn/SiO2催化剂表面酸性中心是TCE脱氯反应的活性中心。Zn/SiO2催化剂在TCE脱HCl反应中存在一定的失活现象,归因于反应过程中催化剂表面积炭。低Zn负载量催化剂上会产生较多积炭,归因于其具有较多强酸性中心,表明催化剂表面强酸中心是导致催化剂积炭和失活的主要原因。
过渡金属催化剂;1,1,2-三氯乙烷;顺-1,2-二氯乙烯;脱氯化氢;酸性;失活
1 引言
在过去的几十年里,氯代有机物在工业界和人们日常生活中广泛使用,如二氯甲烷(CH2Cl2)、三氯乙稀(C2HCl3)、氯苯(C6H5Cl)、多氯二苯并二噁英(PCDDs)等1,但氯代有机物本身具有毒性、致癌和致畸性,在使用过程中会残存于水中、土壤中以及微生物的组织中,对生物体乃至整个生态系统产生严重危害2,3。因此,如何高效处理氯代有机物是一个重要的环保和社会问题。文献中报道处理氯代有机物的方法有焚烧4、催化燃烧5,6、生物降解7,8、光催化9,10、加氢脱氯11,12和脱氯化氢13等。其中脱HCl反应能够将氯代有机物转化为有用的烯烃类物质,是一种高效的废物再利用的办法。本研究将以1,1,2-三氯乙烷(TCE)14为多氯碳氢化合物的典型代表,考察气相催化裂解TCE脱氯化氢生成烯烃反应。
针对气相催化裂解TCE脱HCl反应,目前研究普遍采用碱性较强的催化剂,如含氮有机物催化剂和碱金属盐催化剂,产物是合成偏二氯乙烯(VDC)。Mochida等15利用硅胶负载1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)气相催化TCE脱HCl,得到VDC选择性90%,但TCE稳态转化率非常低(5%)。Serhuchev等16将负载在活性炭上的苯并咪唑作为催化剂进行TCE气相催化脱HCl,得到VDC收率为30%-50%。靳燕霞等17用碱金属CsNO3/SiO2催化剂,在400°C以上反应时,得到TCE转化率98%,VDC选择性78%。
TCE脱HCl主要有三种产物:偏二氯乙烯(VDC)18,顺-1,2-二氯乙烯(cis-DCE)19-21,反-1,2二氯乙烯(trans-DCE)13,目前对合成后两种的1,2-二氯乙烯研究文献较少。DeJournett等22利用卟啉化合物催化剂催化TCE脱HCl,得到cis-DCE和trans-DCE的混合物,且生产cis-DCE的选择性很低。汤岑等23用SiO2负载Mg(NO3)2·6H2O和MgCl2· 6H2O制备MgO/SiO2,在反应温度为300°C时,TCE脱HCl的转化率能够达到92%,同时具有高cis-DCE的选择性(91%),并指出Mg(OH)Cl是TCE脱HCl选择性生成cis-DCE的活性中心。催化剂酸碱性能对TCE脱氯产物有较大影响,如果能够设计和调变催化剂酸碱性,将得到可调变的产物组成,但目前还没有相关文献报道。
第四周期过渡金属催化剂具有较宽范围的酸碱可调性,在多相催化方面有较多应用,如CuO/ Al2O3和NiO/Al2O3催化氧化乙醇和丙酮24,MnOx/ TiO2催化燃烧甲苯25,Mn-Co/TiO2催化氧化NO26。在文献报道中,过渡金属脱氯反应的报道很少,还没发现负载型过渡金属气相催化裂解TCE脱HCl的报道。Baran等27制备了四乙胺BEA分子筛负载Ni催化剂,对1,2-二氯乙烷脱HCl制备氯乙烯进行了研究,发现反应的活性主要是由催化剂的酸性决定,但是反应的活性不高且容易失活。Srebowata等28利用浸渍法制备了四乙胺BEA分子筛负载Co催化剂,用于较低温度下对1,2-二氯乙烷脱HCl制备氯乙烯反应,氯乙烯选择性能够达到90%以上,但反应转化率不高。Srebowata等也研究了相关脱氯反应机理,指出了催化剂酸碱性对催化剂活性和稳定性很重要。
为了考察催化剂酸性对TCE脱氯产物的影响,本研究将在前期考察碱性较强的催化剂17和Mg基催化剂23基础上,用浸渍法制备一系列第四周期的过渡金属催化剂,用于气相催化裂解TCE脱HCl的研究;通过考察催化剂表面酸碱性,探讨过渡金属脱氯作用机理,解释催化剂失活的原因。
2 实验部分
2.1 催化剂制备
1,1,2-三氯乙烷(99.8%)购自上海阿拉丁生化科技股份有限公司;Cr(NO3)3·9H2O、Fe(NO3)3·9H2O、Co(NO3)2·6H2O、Ni(NO3)2·9H2O、Cu(NO3)2· 9H2O、Zn(NO3)2·6H2O购自国药集团化学试剂有限公司;Mn(NO3)2水溶液50%(w)购自上海展云化工有限公司;载体SiO2从青岛美高集团有限公司购买(比表面积379 m2·g-1,孔容积1.02 cm3·g-1,平均孔径15 nm)。
采用浸渍法制备质量分数为10%过渡金属催化剂,标记为10%M/SiO2(M为第IV周期过渡金属)。过渡金属质量分数以硝酸盐前驱体计算,如10%Zn/SiO2,m(Zn(NO3)2)/(m(Zn(NO3)2)+m(SiO2)= 10%)。以10%Zn/SiO2催化剂为例,说明负载型过渡金属催化剂的制备过程:取0.523 g的Zn(NO3)2· 6H2O溶于去离子水,加入3.00 g SiO2,超声0.5 h,在室温下浸渍6 h,100°C水浴炒干,之后置于烘箱中120°C烘干12 h,得到白色固体粉末,Zn负载量为10%,标记为10%Zn/SiO2。
不同Zn负载量的Zn/SiO2催化剂制备过程如下:分别称取0.069、0.250、0.523和1.180 g的Zn(NO3)2·6H2O溶于去离子水,加入3.00 g SiO2,超声0.5 h,在室温下浸渍6 h,100°C水浴炒干,之后置于烘箱中120°C烘干12 h,得到白色固体粉末,其中Zn负载量分别为2%、5%、10%和20%,分别标记为2%Zn/SiO2、5%Zn/SiO2、10%Zn/ SiO2和20%Zn/SiO2。作为对照实验,以氯化锌为前驱体,采用同样的方法制备Zn负载量为10%的催化剂,标记为10%Zn(Cl)/SiO2。
2.2 催化剂表征
热分析表征(TGA)在德国Netzsch STA 449C仪器上进行。空气气氛条件,20 mL·min-1,从室温升温至800°C,升温速率10°C·min-1。
X射线衍射(XRD)表征在荷兰PANalytical公司的X′Pert PROMPD型X射线粉末衍射仪上进行,Cu Ka辐射源,λ=0.1542 nm。仪器的管电压40 kV,管电流40 mA,扫描步长0.014°,扫描范围10°-90°。样品测试是在室温状态下,空气气氛中进行。
物理吸附(BET)表征采用美国Quantachrome公司的Autosorb-1型物理吸附仪测定,催化剂在真空中120°C条件下预处理6 h,-196°C吸附N2。
拉曼光谱在英国Renishaw公司的Invia光谱仪进行,催化剂压片制样,激发激光波长325 nm,每个激光线保持约3 mW的功率,光谱仪的分辨率为1 cm-1,波数范围为200-1800 cm-1。
SEM采用日本Hitachi公司的S4800型扫描电子显微镜(SEM)对吸附剂样品的形貌进行了表征,样品需要经过喷金对其表面进行导电性处理。TEM在工作电压200 kV的JEOL-2100F仪器上进行透射电子显微镜实验。
XPS实验在ESCALAB 250Xi型X射线光电子能谱仪上进行。激发源为单色化Al KαX射线,能量为1486.6 eV,电压为20 kV。分析时的基础真空约为2×10-7Pa。结合能通过表面沉积碳来校正,污染碳的结合能为C l s=284.6 eV,表面元素的分析采用XPSPEAK41软件进行分峰拟合。进而得知催化剂表面离子的种类、对应的峰位置和峰面积。
采用自制NH3-TPD设备表征催化剂表面酸性,实验操作步骤如下:将500 mg催化剂用N2(20 mL·min-1)在500°C下预处理1.5 h,之后冷却至50°C吸附NH3气体0.5 h(20 mL·min-1),然后升温至60°C通N2(20 mL·min-1)吹扫0.5 h,用来除去催化剂表面物理吸附的NH3。接下来以10°C· min-1的升温速率,由60°C升温到800°C,用TCD来检测和记录NH3在升温过程中的脱附信号变化。
2.3 催化剂活性评价
在自制的微型固定床反应装置上进行催化剂评价。典型的评价过程如下:取0.30 g催化剂置于石英反应管中(i.d.=8 mm),热电偶放置于石英管内催化剂的中心位置,以控制反应温度。催化剂先在N2气氛中经过500°C预处理1.5 h,N2气体流速为10 mL·min-1。N2预处理后,使催化剂床层降至一定的反应温度,进行气相裂解TCE脱HCl反应。TCE置于被冰浴冷却的饱和器中,由N2将TCE带入催化剂床层,N2流速为10-30 mL· min-1,对应的空速为1000-3000 h-1。反应产物用安捷伦6890型气相色谱仪(FID)分析,DB-1毛细管柱,30 m×0.25μm。
3 实验结果与讨论
3.1 负载型过渡金属催化裂解TCE脱HCl
图1是负载型过渡金属催化剂上气相裂解TCE脱HCl反应的结果,所有过渡金属负载量为10%,反应温度为350°C。从图1(a)可以看出,在TCE脱HCl反应初期,10%Zn/SiO2、10%Fe/SiO2和10% Co/SiO2催化剂具有较高的TCE转化率(ca.95%),10%Cr/SiO2,10%Ni/SiO2和10%Cu/SiO2催化剂活性较低(转化率小于60%),且载体SiO2活性很低(ca.10%);随着TCE脱HCl反应进行,大部分负载型过渡金属催化剂呈现明显失活现象,而10%Zn/ SiO2催化剂没有明显失活发生,可能原因是由于其他催化剂在反应过程中容易发生中毒现象。在TCE裂解产物中,cis-DCE为主要产物(65%-85%,图1(b)),trans-DCE为主要副产物(15%-30%,图1(c))。随着脱氯反应的进行,大部分过渡金属催化剂上cis-DCE选择性呈现缓慢下降趋势(10%Fe/SiO2除外)。通过对M/SiO2催化剂的性能比较,发现10%Zn/SiO2催化剂能得到较好的催化性能,TCE转化率98%,cis-DCE选择性83%,因此本研究主要围绕Zn/SiO2催化剂展开。
3.2 Zn/SiO2催化裂解TCE脱HCl
图2是Zn前驱体(Zn(NO3)2和ZnCl2)对Zn/SiO2催化剂气相催化裂解TCE脱HCl反应结果。从图2(a)可以看出,以Zn(NO3)2为前驱体得到的Zn/SiO2催化剂,在TCE脱HCl反应过程中,TCE转化率和各种产物选择性基本保持稳定:TCE转化率98.1%,cis-DCE和trans-DCE选择性分别为81.5%和15.1%,氯乙炔等其他副产物(others)总和小于5%。从图2(b)可以看出,由ZnCl2为前驱体得到的Zn(Cl)/SiO2催化剂,其催化性能与Zn(NO3)2前驱体的非常一致,表明两种前驱体对Zn/SiO2催化剂上TCE脱HCl反应的影响较小,可能原因是在脱HCl过程中,二者都转变为相同Zn的氯化物23。本研究主要以Zn(NO3)2为前驱体制备Zn/SiO2催化剂,考察预处理温度、反应温度、反应空速、Zn负载量和催化剂用量等因素影响,以获得较好的多氯代烷烃脱HCl的催化性能。
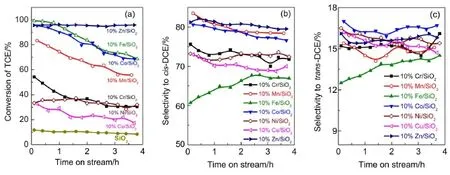
图1 负载型过渡金属催化剂上气相裂解1,1,2-三氯乙烷(TCE)脱HCl反应Fig.1 Dehydrochlorination of 1,1,2-trichloroethane(TCE)catalyzed by SiO2-supported transition metals (a)TCE conversion;(b)selectivity to cis-1,2-dichloroethylene(cis-DCE);(c)selectivity to trans-1,2-dichloroethylene(trans-DCE); reaction condition:amountof catalyst,0.30 g;pretreatmentcondition:500°C in N2for 1.5 h;reaction temperature:350°C;space velocity:1000 h-1
表1是预处理温度、反应温度和空速等对Zn/ SiO2催化剂上气相裂解TCE脱HCl反应的影响结果,所有数据来自反应进行2 h时采样分析。从表1中可以看出,在预处理温度400-600°C范围内,预处理温度对TCE转化率(ca.97%)和各种产物选择性影响较小。
反应温度对TCE转化率影响较大,300°C以下得到较低TCE转化率(ca.60%),350°C以上能够得到TCE转化率98.1%,再增加反应温度没有明显变化。反应温度对产物选择性也有明显影响:cis-DCE选择性随着反应温度升高而降低,而trans-DCE选择性随着反应温度升高而增加,这表明高温反应(400°C以上)有利于trans-DCE生成。
随着空速增加,TCE转化率逐渐降低。1000 h-1以下低空速具有高稳定的TCE转化率(ca.98%,图2(a)),当采用3000 h-1以上较高空速,TCE转化率在反应过程中会逐渐下降。改变空速对产物cis-DCE选择性影响较小(ca.82%)。综合以上因素,较优Zn/SiO2催化剂的评价工艺为:预处理温度500°C,反应温度350°C,空速1000 h-1。
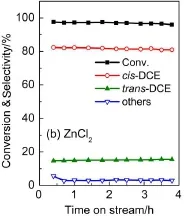
图2 Zn前驱体对Zn/SiO2催化剂上气相裂解TCE脱HCl反应的影响Fig.2 Effect of Zn precursor(Zn(NO3)2or ZnCl2)on the dehydrochlorination of 1,1,2-trichloroethane(TCE)over 10%Zn/SiO2(a)Zn(NO3)2;(b)ZnCl2.reaction condition:amountofcatalyst,0.30 g;pretreatmentcondition,500°C in N2for 1.5 h; reaction temperature,350°C;space velocity:1000 h-1
图3 是Zn负载量(2%-20%)对气相裂解TCE脱HCl反应的影响结果。从图3(a)可以看出,Zn负载量对TCE初始转化率具有较大影响,10%以上Zn负载量达到98%以上,2%以下Zn负载量低于65%。随着脱氯反应进行,10%以上的较高负载量催化剂上TCE转化率基本保持稳定(>98%),而5%以下的较低负载量催化剂存在显著失活过程。从图3(b)中可以看出,随着Zn负载量的增加,生成cis-DCE的选择性逐渐升高。较高Zn负载量具有较好的催化性能,可能与其具有较多催化活性中心,以及催化剂表面的酸性强度等有关,下面会做详细阐述。
需要引起注意的是,当Zn负载量达到10%以上时(图3(a)),TCE脱HCl转化率均达到98%以上,在实验条件下没有明显失活,表现出高稳定性的催化活性。引起这个现象可能原因有两个:一是5%以下Zn负载量催化剂容易失活;二是10%以上Zn负载量催化剂活性位多,而评价反应时间不够长,造成实验条件下没有失活的假象。本研究首先采用减少催化剂用量,检验10%以上Zn负载量的催化剂是否真实高效稳定。
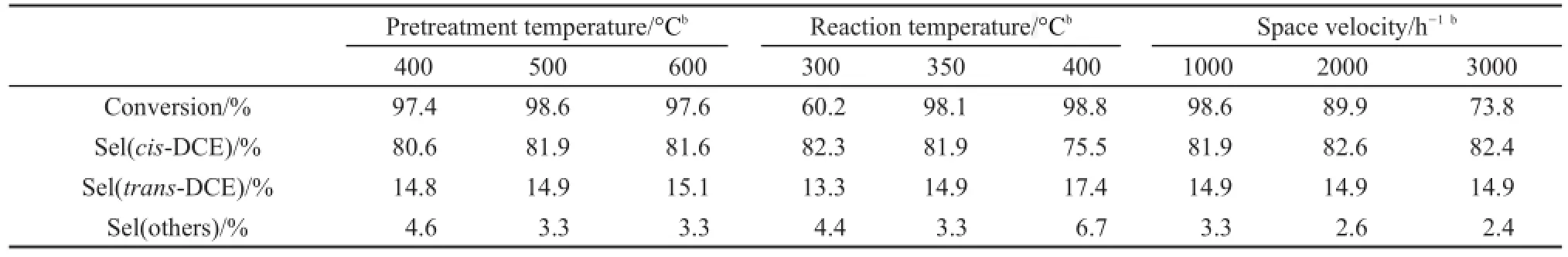
表1 预处理温度、反应温度和空速对10%Zn/SiO2催化剂上TCE气相裂解反应的影响Table 1 Effect of pretreatment temperature,reaction temperature and space velocity on the dehydrochlorination of TCE over 10%Zn/SiO2a

图3 Zn负载量对Zn/SiO2催化剂上TCE气相裂解反应的影响Fig.3 Effect of Zn loadings on the dehydrochlorination of TCE over Zn/SiO2catalysts (a)TCE conversion;(b)selectivity to cis-DCE.reaction condition:amountofcatalyst,0.30 g; pretreatmentcondition:500°C in N2for 1.5 h;reaction temperature,350°C;space velocity:1000 h-1
将Zn/SiO2催化剂用量由0.3 g减少到0.1 g,同时采用空白载体补充以保持相同体积,所得到的气相裂解TCE脱HCl结果见图4。从图4(a)可以看出,在减少催化剂用量后,10%负载量时TCE初始转化率为88%,明显低于20%负载量(98%)。因此,10%以上Zn负载量在实验条件下,能够表现出较稳定催化活性,应该归因于催化活性位总量较多,且评价反应时间不够长。10%以上高负载量催化剂都呈现明显失活过程(图4(a)),这是催化剂用量较多时被掩盖的本质现象。
将不同负载量Zn/SiO2催化剂的转化率换算成比活性(单位催化剂质量和单位时间内转化TCE的物质的量),所得结果见图4(b)。从图4(b)中可以看出,随着Zn负载量的增加,Zn/SiO2催化剂的比活性是呈现显著下降趋势,与表观反应活性恰好相反(图3(a)和4(a))。低负载量催化剂具有较高比活性,可能与其催化剂表面酸性化学环境有关。此外,减少催化剂用量,对cis-DCE选择性的影响不大(图4(c))。
3.3 Zn/SiO2催化剂失活原因
上述实验结果表明(图4(a)),不同Zn负载量的Zn/SiO2催化剂在脱氯过程中均存在失活的本质现象。引起催化剂失活原因有多种29,其中常见的是催化剂活性中心物种晶粒尺寸变大,分散度降低,或催化剂活性中心被覆盖,如积炭等。以下各种表征是针对这些可能原因的探究。
XRD和BET表征是揭示催化剂活性物种晶粒尺寸及分散度的有效手段,因此本研究对反应前后的10%Zn/SiO2催化剂进行了XRD和BET表征,结果见图5。从图5可以发现,10%Zn/SiO2催化剂反应前后的衍射峰均与空白载体SiO2一致,没有出现明显Zn物种衍射峰,表明在脱氯反应发生后,Zn物种没有颗粒聚集而产生较大晶粒。BET表征得到催化剂反应前后的比表面积没有发生明显变化(ca.300 m2·g-1),表明脱氯反应没有影响Zn/SiO2催化剂孔道结构,结合XRD表征结果,可以得出Zn物种SiO2载体催化剂上是高分散的。
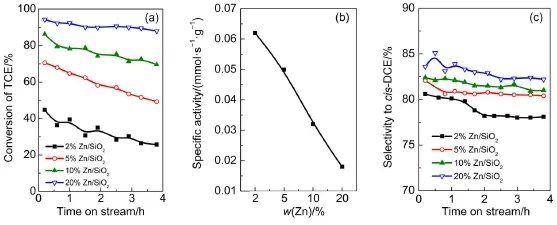
图4 Zn/SiO2催化剂用量减少后(0.10 g)TCE气相裂解反应Fig.4 Dehydrochlorination of TCE over Zn/SiO2catalysts after decreasing the amount of Zn catalysts (a)TCE conversion;(b)specific activity;(c)selectivity to cis-DCE.reaction condition:amountofcatalyst,0.10 g;pretreatmentcondition, 500°C in N2for 1.5 h;reaction temperature:350°C;space velocity:1000 h-1

图5 SiO2和10%Zn/SiO2催化剂的XRD和BET表征Fig.5 XRD and BET characterization of SiO2and 10%Zn/SiO2catalysts
为了进一步考察TCE脱氯反应对Zn/SiO2催化剂物种颗粒的影响,采用电镜技术表征了反应前后10%Zn/SiO2催化剂(图6)。从图6中可以看出,无论是表面分析SEM,还是内部分析TEM,都表明反应前后物种颗粒基本一致,因此,关于Zn/ SiO2催化剂活性在脱氯过程中降低,基本排除Zn物种晶粒尺寸增大和分散度降低等因素。
催化剂活性物种在反应中变化,也可能是导致催化剂活性降低的一个主要原因,因此,采用XPS表征反应前后Zn/SiO2催化剂,以考察脱氯过程中Zn物种变化,结果如图7所示。从图7中可以看出,低负载量的催化剂在反应之前,主要表现为ZnO特征峰30,31,反应之后出现ZnCl2特征峰30,表明在脱氯过程中,ZnO逐渐被脱附出的HCl氯化而转变为ZnCl2。结合不同Zn前驱体的实验结果,ZnCl2前驱体具有较好的催化性能(图2),因此,在脱氯过程中产生ZnCl2不会导致催化剂失活,催化剂失活可能与催化剂表面积炭有关。
多相催化反应中催化剂表面积炭,覆盖催化剂活性中心是导致催化剂失活的重要原因32。表征催化剂表面积炭的常用手段有TGA和拉曼(Raman)等表征。图8是10%Zn/SiO2催化剂在反应前后的TGA表征结果。从图8可以看出,新鲜10%Zn/ SiO2催化剂在100°C以下有一个明显失重过程(ca. 4%),归属于催化剂表面吸附水的脱附。反应后10%Zn/SiO2催化剂有两个明显的失重过程,其一是100°C以下有一个失重过程(ca.8%),归属于催化剂表面吸附水的脱附,以及cis-DCE等小分子脱附(沸点低于100°C)17,其二是410°C以上有一个失重过程(ca.8%),归属于积炭或含碳有机聚合物的燃烧失重。
为了验证催化剂表面是否存在积炭,本研究对反应后的Zn/SiO2催化剂进行了拉曼表征,结果见图9(a)。从图9(a)可以看出,反应后不同负载量的Zn/SiO2催化剂均存在明显的拉曼衍射峰(1300和1600 cm-1),归属于催化剂表面的积炭20,这表明Zn/SiO2催化剂在脱氯过程中产生积炭。由于不同负载量的Zn/SiO2催化剂都存在失活现象(图4(a)),因此,可以推测催化剂失活主要由积炭引起,即积炭覆盖了催化剂活性中心。

图6 10%Zn/SiO2催化剂的SEM(a,b)和TEM(c,d)表征Fig.6 SEM(a,b)andTEM(c,d)characterization for fresh and used 10%Zn/SiO2catalysts

图7 5%Zn/SiO2催化剂和10%Zn/SiO2催化剂的XPS表征Fig.7 High-resolution XPS data of the Zn 2p3/2peak for the fresh and used samples of 5%Zn/SiO2and 10%Zn/SiO2catalysts

图8 反应前后10%Zn/SiO2催化剂的TGA表征Fig.8 Thermo gravimetric analysis(TGA)curve of fresh and used 10%Zn/SiO2catalysts in Air atmosphere
根据对不同负载量的XRD和BET的表征结果,20%以下Zn负载量时,Zn物种在载体上基本是高分散的,因而可以认为多数Zn物种是参与TCE脱HCl反应的,将各种催化剂表面积炭的拉曼峰面积除以Zn负载量,得到不同负载量催化剂的归一化积炭量(比积炭量),见图9(b)。从图9(b)中可以看出,随着Zn负载量增加,Zn/SiO2催化剂表面的比积炭量是逐渐降低,这表明高负载量催化剂不容易被积炭覆盖活性中心,从而表现出较高催化活性(图3(a)和4(a))。低负载量催化剂表面积炭量较多,尤其2%Zn/SiO2催化剂,其比积炭量是20%Zn/SiO2催化剂的9.5倍。不同负载量催化剂上的积炭差异,可能与其表面酸碱度有关,因此下面进行催化剂表面酸碱性研究(NH3-TPD表征)。
图10是不同负载量Zn/SiO2催化剂的NH3-TPD表征结果,催化剂经过N2气氛中500°C预处理。从10(a)图可以看出,不同负载量Zn/SiO2催化剂上均出现两个NH3脱附峰,分别归属于弱酸中心(α峰)和强酸中心(β峰)。随着Zn负载量的增加,弱酸中心(α和β峰)的峰面积呈现增加趋势,表明较高Zn负载量的催化剂具有较多酸量。随着Zn负载量的增加,弱酸中心(α峰)和强酸中心(β峰)都向低温方向偏移,其中强酸中心(β峰)偏移非常明显。催化剂酸中心向低温方向偏移,表明表面酸强度逐渐减弱,由此得出低负载量的催化剂具有较强的酸性中心33。
将图10(a)中弱酸性峰面积(α峰)除以相应催化剂的Zn负载量,归一化得到各种催化剂的比酸性量,见图10(b)。从图10(b)中可以看出,高负载量具有较少比酸性量,低负载量催化剂具有较多比酸性量。

图9 反应后Zn/SiO2催化剂的Raman表征(a)和归一化积炭量(b)Fig.9 Raman spectroscopy(a)and normalized surface carbon deposit(b)of used 10%Zn/SiO2 catalysts with different Zn loadings

图10 不同Zn负载量的Zn/SiO2催化剂NH3-TPD表征(a)和比酸量(b)Fig.10 NH3Temperature Programmed Desorption (NH3-TPD)(a)and normalized surface acidity(b)of Zn/SiO2catalysts with different Zn loadings

图11 Zn/SiO2催化剂上脱氯活性中心和失活机理示意图Fig.11 Possible activity center and deactivation mechanism for the dehydorchlorination of TCE over Zn/SiO2catalysts
图11 给出了Zn/SiO2催化剂上可能的脱氯活性中心和失活机理示意图。随着Zn负载量增加,催化剂表面酸量逐渐增加(图10(a)),表观催化活性显著提高(图4(a)),这表明催化剂表面酸中心是催化剂活性中心。低负载量催化剂虽然表观催化活性低,但其具有相对多的比酸性量(图10(b)),而且具有较高的比活性(图4(b)),进一步说明催化剂表面酸性位越多,催化剂活性越高,由此得出,在Zn/ SiO2催化TCE脱氯化氢过程中,脱氯活性中心为Zn物种引起的催化剂表面酸中心。
Zn/SiO2催化剂在TCE脱氯反应中存在失活现象(图4(a)),催化剂失活主要是积炭引起(图9(a)),那么是什么原因引起积炭,或加速积炭呢?通过对比不同负载量Zn催化剂的酸性强度(图10(a)),可以发现随着Zn负载量的增加,Zn催化剂酸中心向低温方向偏移,低负载量的催化剂具有较强的酸性中心。通过比较图3(a)和图4(a)可以发现,随着Zn负载量减少,催化剂失活越明显,说明催化剂酸性越强就越容易积炭失活。由于低负载量催化剂上比积炭量较大(图9(b)),因此可以得出,催化剂表面强酸中心是导致积炭,并引起失活的主要因素。
4 结论
相对于多数SiO2负载的第四周期过渡金属催化剂,Zn/SiO2对气相裂解TCE脱HCl具有较高转化率(98%)和cis-DCE的选择性(83%)。不同Zn负载量的Zn/SiO2催化剂具有不同催化活性,其本质与催化剂酸量有关。研究发现Zn/SiO2催化剂中比酸量越大比活性越高,表明Zn/SiO2催化剂表面酸性中心是TCE脱氯反应的活性中心。不同Zn负载量催化剂均存在失活现象,主要原因是催化剂表面的积炭造成的。当Zn/SiO2催化剂具有较强的酸性中心时,具有较大比积炭量,表明催化剂表面强酸中心是导致积炭,从而导致催化剂失活的主要原因。至于Zn/SiO2催化剂上强、弱酸中心的具体形成原因,需要进行下一步深入的研究。
(1)Wang,M.X.;Liu,S.T.Chem.Ind.Eng.2015,32,3[王明玺,刘善堂.化学工业与工程,2015,32,3.]doi:10.13353/j. issn.1004.9533.2015.03.005
(2)Zhang,B.Study on Chlorine Species ofAir Particles in Shijingshan.MS Dissertation,Chengdu University of Technology,Chengdu,2013.[张博.北京市石景山区大气环境中氯种态研究[D].成都:成都理工大学,2013.]
(3)Wei,M.Photocatalytic Reductive dechlorination of chlorophenols by TiO2catalysts.Ph D Dissertation,Hubei University of Technology,Wuhan,2016.[魏蒙.TiO2光催化剂对氯酚化合物的还原脱氯性能的研究[D].武汉:湖北工业大学,2016.]
(4)Stach,J.;Pekáarek,V.;Endrst,R.;Hetflejs,J.Chemosphere 1999,39,2391.doi:10.1016/S0045-6535(99)00166-6
(5)Coute,N.;Ortego Jr,J.D.;Richardson,J.T.;Twigg,M.V.Appl. Catal.B-Environ 1998,19,175.doi:10.1016/S0926-3373(98) 00075-7
(6)Rivas,B.;Sampedro,C.;López-Fonseca,R.;Gutiérrez-Ortiz, M.Á.;Gutiérrez-Ortiz,J.I.Appl.Catal.A:Gen.2012,417,93. doi:10.1016/j.apcata.2011.12.028
(7)Barriault,D.;Sylvestre,M.Can.J.Microbiol.1993,39,594. doi:10.1139/m93-086
(8)Yu,J.;Cai,W.;Zhao,S.;Wang,Y.;Chen,J.Chin.J.Chem.Eng. 2013,21,781.doi:10.1016/S1004-9541(13)60536-4
(9)Trillas,M.;Peral,J.;Domènech,X.J.Chem.Tech.Biotol.1996, 67,237.doi:10.1002/(SICI)1097-4660(199611)67:3<237::AIDJCTB567>3.0.CO;2-4
(10)Ahmed,S.;Ollis,D.F.S ol.Energy 1984,32,597.doi:10.1016/ 0038-092X(84)90135-X
(11)Park,Y.;Kang,T.;Cho,Y.S.;Kim,P.;Park,J.C.;Yi,J.Stud. Surf.Sci.Catal.2003,146,637.doi:10.1016/S0167-2991(03) 80464-0
(12)Gampine,A.;Eyman,D.P.J.Catal.1998,179,315. doi:10.1006/jcat.1998.2223
(13)Alwies,W.A.M.van der Heijden;Ad,J.M.Mens;René, Bogerd;Bert,M.Weckhuysen.Catal.Lett.2008,122,238. doi:10.1007/s10562-008-9436-2
(14)Bartsch,R.;Curlin,C.L.;Florkiewicz,T.F.;Minz,H.R.; Navin,T.;Scannell,R.;Zelfel,E.Chlorine:Principles and Industrial Practice;Wiley-VCH GmbH:Weinheim,2000.
(15)Mochida,I.;Watanabe,H.;Uchino,A.;Fujitsu,H.;Takeshita, K.;Furuno,M.;Sakura,T.;Nakajima,H.J.Mol.Catal.1981, 12,359.doi:10.1016/0304-5102(81)85040-7
(16)Serhuchev,Y.O.;Bilokopytov,Y.V.;Chernobaev,I. Composition for the Vapor Phase Dehydrohalogenation of 1,1, 2-Trihaloethane to 1,1-Dihaloethylene and Methods for Preparing and Using Such Composition.US 2008242902 A1, 2008.
(17)Jin,Y.X.;Tang,C.;Meng,X.Q.;Wang,X.X.;Xie,G.Q.;Luo, M.F.;Li,X.N.Acta Phys.-Chim.Sin.2016,32,510.[靳燕霞,汤岑,孟秀清,王小霞,谢冠群,罗孟飞,李小年.物理化学学报,2016,32,510.]doi:10.3866/PKU.WHXB201511134
(18)Kapp,R.W.Vinyl Chloride A2.In Encyclopedia ofToxicology, 3rd ed.;Wexler,P.Ed.;Academic Press:Oxford,2014;pp 934-938.
(19)Dolfing,J.;Janssen,D.Biodegradation 1994,5,21. doi:10.1007/BF00695210
(20)Kokubo,K.;Kitasaka,K.;Oshima,T.Org.Lett.2006,8,1597. doi:10.1021/ol060198d
(21)Turton,D.A.;Martin,D.F.;Wynne,K.Phys.Chem.Chem. Phys.2010,12,4191.doi:10.1039/B918196B
(22)DeJournett,T.D.;Fritsch,J.M.;McNeill,K.;Arnold,W.A.J. Labelled Comp.Radiophram.2005,48,353.doi:10.1002/ jlcr.929
(23)Tang,C.;Jin,Y.X.;Lu,J.Q.;Li,X.N.;Xie,G,Q.;Luo,M.F. Appl.Catal A-Gen.2015,508,10.doi:10.1016/j. apcata.2015.09.024
(24)Guo,J.G.;Li,Z.;Xi,H.X.;He,Y.S.;Wang,B.G.China Univ. Tech.(Natural.Sci.Edit.)2004,32,5.[郭建光,李忠,奚红霞,何余生,王伯光.华南理工大学学报(自然科学版),2004, 32,5.]
(25)Qi,L.L.;Yao,J.;You,H.J.Harb.Inst.Technol.2010,42,6.[亓丽丽,姚杰,尤宏.哈尔滨工业大学学报,2010,42,6.]
(26)Zhao,J.;Wang,H.R.;Zhu,T.Y.;Li,P.;Jing,P.F.Acta Phys.-Chim.Sin.2013,29,385.[赵俊,王海蕊,朱廷钮,李鹏,荆鹏飞.物理化学学报,2013,29,385.]doi:10.3866/ PKU.WHXB201212031
(27)Baran,R.;Srebowata,A.;Kaminska,I.I.;Łomot,D.;Dzwigaj, S.Micropor.Mesopor.Mat.2013,180,209.doi:10.1016/j. micromeso.2013.06.037
(28)Anna,S.B.;Rafal,B.;Izabela.I.K.;Thomas,O.;Jean,M.K.; Stanislaw,D.Catal.Today 2015,251,73.doi:10.1016/j. cattod.2014.10.019
(29)Chen,X.Z.;Cui,B.;Shi,W.P.;Luo,M.Y.I nd.Catal.2001,9, 5[陈晓珍,崔波,石文平,罗明焰.工业催化,2001,9,5.]
(30)Ali,H.A.;Iliadis,A.;Lee,U.Solid State Electron 2004,48, 2025.doi:10.1016/j.sse.2004.05.052
(31)Ali,H.A.;Iliadis,A.Thin Solid Films 2005,471,145. doi:10.1016/j.tsf.2004.05.074
(32)Rossetti,I.;Bencini,E.;Trentini,L.Appl.Catal.A-Gen.2005, 292,118.doi:10.1016/j.apcata.2005.05.046
(33)Guimon,C.;Auroux,A,;Romero,E.Appl.Catal.A-Gen.2003, 251,199.doi:10.1016/S0926-860X(03)00318-1
Gas Phase Dehydrochlorination of 1,1,2-Trichloroethane over Zn/SiO2Catalysts:Acidity and Deactivation
HU Yi-Hao SONGTong-Yang WANGYue-Juan HU Geng-Sheng XIE Guan-Qun*LUOMeng-Fei*
(Key Laboratory of the Ministry of Education for Advanced Catalysis Materials,Institute of Physical Chemistry, Zhejiang Normal University,Jinhua 321004,Zhejiang Province,P.R.China)
A series of SiO2-supported fourth period transition metalcatalysts(M/SiO2)was prepared by a wetness impregnation method for the dehydrochlorination of 1,1,2-trichloroethane(TCE)in the gas phase. Among these M/SiO2catalysts,Zn/SiO2had the bestcatalytic activity with the highest TCE conversion(~98%) and excellentselectivity for cis-1,2-dichloroethylene(82%).By increasing the zinc loading,the conversion of TCE using the Zn/SiO2catalyst was gradually improved,in agreement with the totalacidity in the Zn/SiO2catalyst.Associating the specific activity and specific acidity ofthe Zn/SiO2catalystwith different Zn loadings, itwas found thathigher specific acidity contributed to higher specific activity,indicating thatthe acid center of Zn/SiO2was the catalytic active site for the dehydrochlorination of TCE.In the process ofdehydrochlorination, the Zn/SiO2catalystcould be deactivated,mainly due to coke deposition on the catalystsurface.Catalysts with low Zn loading had stronger acid sites,which resulted in more coke formation on the catalyst.The results showed thatstrong acid sites on the catalystsurface were responsible for the deposition ofcoke and deactivation ofthe catalyst.
Transition metalcatalysts;1,1,2-Trichloroethane;Cis-1,2-dichloroethylene; Dehydrochlorination;Acidity;Deactivation
O643
10.3866/PKU.WHXB201702082
Received:November 25,2016;Revised:January 23,2017;Published online:February 8,2017.
*Corresponding authors.XIE Guan-Qun,Email:gqxie@zjnu.edu.cn.LUO Meng-Fei,Email:mengfeiluo@zjnu.edu.cn. The projectwas supported by the Natural Science Foundation of Zhejiang Province,China(LY16B070001).
浙江省自然科学基金(LY16B070001)资助项目©Editorialoffice ofActa Physico-Chimica Sinica
猜你喜欢
化学工程师(2023年1期)2023-02-17 15:09:48
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
中国果树(2020年2期)2020-07-25 02:14:28
上海农业科技(2019年1期)2019-02-22 01:51:28
汽车维护与修理(2018年7期)2018-10-13 06:03:56
石油学报(石油加工)(2015年6期)2015-07-02 01:39:44
化学反应工程与工艺(2015年1期)2015-04-16 03:06:12
河南科技(2015年2期)2015-02-27 14:20:35
燃气轮机技术(2014年4期)2014-04-16 03:54:03
食品科学(2013年19期)2013-03-11 18:27:17
