
基于P—糖蛋白的肠吸收在体单向肠灌流模型验证
2017-05-11 11:21董月柳刘洋尹秀文潘孟李雪莲王子
中国中药杂志 2017年8期
董月柳+刘洋+尹秀文+潘孟+李雪莲+王子禹+董玲

[摘要] 对基于P-糖蛋白(P-gp)肠吸收单灌流模型进行验证。首先,通过酚红灌流,采用质量法进行水分校正,通过高效液相色谱法对灌流前后酚红水平进行检测,以验证肠上皮细胞的紧密连接结构正常,且肠上皮的完整性保持良好;其次,采用FDA指定阳性药地高辛对模型进行验证,给予大鼠不同质量浓度维拉帕米后,对大鼠回肠段地高辛吸收参数进行观察对比。酚红在大鼠体内回肠段存在吸收,吸收参数有效渗透系数Peff为(1.09±0.62)×10-6 cm·s-1,结果表明肠上皮细胞的紧密连接结构正常,且肠上皮的完整性保持良好,地高辛灌流不给予维拉帕米时,地高辛在回肠段存在一定程度的吸收,吸收参数有效渗透系数Peff为(1.07±0.59)×10-5 cm·s-1,给予大鼠0.01,0.1 mmol·L-1浓度的维拉帕米后,地高辛在大鼠回肠的吸收呈上升趋势,吸收参数有效渗透系数Peff分别为(1.58±0.69)×10-5,(3.28±0.95)×10-5 cm·s-1,高浓度时,数据差异具有统计学意义(P<0.05)。地高辛灌流实验验证了小肠上皮P-gp表达完整,可以用于P-gp外排转运体的研究。
[关键词] 在体单向肠灌流模型;酚红;地高辛;P-gp
Validation of in situ single pass perfusion model based on P-gp
DONG Yue-liu, LIU Yang, YIN Xiu-wen, PAN Meng, LI Xue-lian, WANG Zi-yu, DONG Ling*
(Beijing University of Chinese Medicine, Beijing 100102, China)
[Abstract] To validate in situ rats intestinal single pass perfusion model based on P-glycoprotein (P-gp). Firstly, phenol red perfusion was carried out to verify the close connection structure of intestinal epithelial cells, and the integrity of the intestinal epithelium, with a gravimetric method for correcting water flux. The level of phenol red was determined by high performance liquid chromatography (HPLC) both before and after perfusion. Secondly, the positive drug digoxin specified by FDA was used to validate the model. After different mass concentrations of verapamil were given in the rats, the absorption parameters of digoxin in ileum of rats were observed and compared. The results showed that the phenol red was absorbed in rats ileum segment, with an effective permeability coefficient of (1.09±0.62)×10-6 cm·s-1. The experiment results indicated that the close connection structure of intestinal epithelial cells was normal, and the integrity of the intestinal epithelium was maintained well. In digoxin perfusion experiment, in case no verapamil was given, digoxin showed certain degree of absorption in rat ileum, with an effective permeability coefficient (Peff) of (1.07±0.59)×10-5 cm·s-1; after mass concentrations of 0.01,0.1 mmol·L-1 verapamil were given, the absorption of digoxin was on the rise in rat ileum, with an effective permeability coefficient Peff of (1.58±0.69)×10-5, (3.28±0.95)×10-5 cm·s-1 respectively (P<0.05). Digoxin perfusion experiment verified that P-gp expression in small intestine epithelium was intact and can be used in the research of P-gp efflux transporter.
[Key words] in situ rat intestinal perfusion; phenol red; digoxin; P-gp
口服是用药方式最常见的,因为它具有方便、安全、低成本和患者依从性的特点[1]。口服吸收取决于药物从剂型中释放和在肠膜的渗透,所以药物的溶解和渗透过程是药物吸收最基本的条件[2]。基于此理论,Amidon等[3]提出了生物药剂学分类系统(biopharmaceutics classification system,BCS),根据药物的溶解度和渗透性高低将药物分为四类。2000年美国 FDA颁布BCS作为1种科学方法用于速释固体口服剂型Ⅰ类(高溶解度和高渗透性)的体内生物利用度豁免和生物等效性试验[4],以此科学框架制订了药品生物等效豁免的相关技术要求,即对于BCSⅠ类的药品上市后的变更、仿制给予免除生物等效性研究的支持。随着BCS的不断深入研究,发现其对已上市药品生物利用度的认识具有重要价值,从而继续前移到药品开发甚至前体药物筛选,将BCS推至口服药物成药性论证的核心地位,通过制剂手段改变药物的BCS分类也成为生物药剂学研究和新剂型研究的重要目标之一。BCS是基于化学药物提出来的,而中药具有多成分的特点,这就决定了研究中药的渗透性和溶解度时要考虑它所处的多成分环境,因此本课题组借鉴化学药物BCS的优势结合中药多成分的特点,提出了中药生物药剂学分类系统[5],课题组前期通过单成分到两成分再到多成分配伍逐渐递增的思路对葛根芩连方中的高含量成分葛根素和小檗碱在多成分环境中的渗透性和溶解度进行研究,研究結果显示,成分与成分之间存在相互作用[6-8],这种相互作用引起药物的肠渗透性的改变。药物的主要吸收部位是小肠,药物在小肠吸收的同时发生着多种过程[9],在人体中低渗透的药物可能是膜上的外排转运体将药物外排引起的,例如P-gp。研究渗透性的模型,美国FDA推荐使用动物的体内、在体灌流模型或体外模型(切除的肠组织或者合适的上皮细胞单层膜)[10-11],通常使用Caco-2细胞模型。然而当外排蛋白在研究模型上存在缺陷或者表达的程度低于人体时,存在外排转运的药物比被动转运的药物更容易出现错误的渗透性分类[12],相关研究结果也显示,FDA中BCS的指导原则中体外细胞渗透模型不能准确预测药物在人体的吸收程度[13-14],所以选择合适的模型研究药物的转运机制至关重要。本课题通过酚红和地高辛灌流实验分别对与人体相似性大的在体单向肠灌流模型的肠上皮细胞的紧密连接结构以及P-gp表达完整性进行验证,有利于更好地为药物进行正确的BCS分类提供条件。
1 材料
1.1 仪器
LC-20AT高效液相色谱仪(SPD-20A型紫外检测器,SIL-20A自动进样器,日本岛津公司);BT-25S电子分析天平(北京赛多利斯仪器有限公司);DZKW-4电热恒温水浴锅(北京中兴伟业仪器有限公司);KH7200DB型数控超声波清洗器(昆山超声仪器有限公司);STARTER2100实验室pH计(奥豪斯仪器上海有限公司)。
1.2 试剂与药品
酚红对照品(批号P0882-25G,上海百赛生物技术有限公司),地高辛对照品(批号20140316,南通飞宇生物科技有限公司),乙腈(色谱级,美国Fisher公司),娃哈哈纯净水购买于娃哈哈集团公司(中国杭州),其他试剂均为分析纯。
1.3 动物
SD大鼠,雄性,体重200~220 g,斯贝福(北京)实验动物技术有限公司提供,许可证号 SCXK(京)2011-0004。
2 方法
2.1 供试液的配制
2.1.1 Krebs-Ringer′s营养液(K-R液)配制
称取NaCl 7.8 g,KCl 0.35 g,CaCl2 0.37 g,NaHCO3 1.37 g,NaH2PO4 0.32 g,MgCl2 0.02 g,葡萄糖1.4 g,加去离子水定容至1 000 mL,即得,加入NaHCO3,调节pH为7.4。
2.1.2 酚红对照品储备液制备
精密称定酚红0.1 g置于50 mL量瓶中,用K-R液定容,得2 g·L-1的酚红母液。
2.1.3 酚红灌流液制备
取2.0 mL母液置于200 mL量瓶中,K-R液稀释并定容得20 mg·L-1的酚红溶液。
2.1.4 地高辛母液制备
精密称定地高辛对照品5 mg,置于50 mL量瓶中,加甲醇溶解并定容,得100 mg·L-1的地高辛母液。
2.1.5 地高辛灌流液制备
取地高辛母液5 mL,置于100 mL量瓶中,用K-R液定容,得5 mg·L-1的地高辛灌流液。
2.2 含量测定方法
2.2.1 酚红含量测定方法
2.2.1.1 酚红色谱条件 GEMINI C18色谱柱(4.6 mm×250 mm,5 μm,Phenomenex),流速1.0 mL·min-1,检测波长230 nm,柱温30 ℃,进样量20 μL,流动相0.1%磷酸(A)-乙腈(B)(68∶32),等度洗脱。
2.2.1.2 标准曲线制备 精密吸取酚红对照品储备液,以适量的K-R 缓冲液 (pH 7.4) 将其分别稀释为2.5,5,10,20,40,80 mg·L-1,进样20 μL,以质量浓度(X,mg·L-1),对峰面积 (Y) 进行线性回归,得标准曲线方程。
2.2.1.3 回收率 用空白肠灌流液配制低、中、高 (2.5,20,80 mg·L-1) 3個浓度的酚红溶液,分别进样,测定峰面积,考察酚红的回收率。
2.2.1.4 精密度 取酚红对照品溶液连续进样6次,测定酚红峰面积。
2.2.1.5 重复性 取低、中、高3个对照品浓度依次进样3次,测定酚红峰面积。
2.2.1.6 稳定性 取酚红供试品高、中、低3个浓度,将供试品溶液置于37 ℃的水浴中3 h,按上述液相色谱条件测定峰面积。
2.2.2 地高辛含量测定
2.2.2.1 地高辛色谱条件 GEMINI C18色谱柱(4.6 mm×250 mm,5 μm,Phenomenex),流动相1% 冰醋酸(A)-乙腈(B)(68∶32),等度洗脱,检测波长230 nm,柱温30 ℃,流速1.0 mL·min-1,进样量20 μL。
2.2.2.2 标准曲线制备 精密吸取地高辛对照品贮备液5 mL置于10 mL量瓶中,加入5 mL K-R 液,将其分别稀释为质量浓度为3.125,6.25,12.5,25,50 mg·L-1系列溶液。以地高辛浓度(X) 对峰面积(Y) 绘制标准曲线,进行线性回归。
2.2.2.3 回收率 用空白肠灌流液配制低、中、高 (2.5,20,80 mg·L-1) 3个浓度的地高辛溶液,分别进样,测定峰面积,考察地高辛的回收率。
2.2.2.4 重复性 取低、中、高3个对照品浓度依次进样3次,测定地高辛峰面积。
2.2.2.5 精密度 取地高辛对照品溶液,连续进样6次,测定地高辛峰面积。
2.2.2.6 稳定性 取地高辛母液1 mL置于20 mL量瓶中,K-R液稀释并定容得5 mg·L-1的灌流液,将灌流液置于37 ℃的水浴中3 h,按上述液相色谱条件测定峰面积。
2.3 大鼠在体肠吸收试验
大鼠禁食不禁水18 h,水合氯醛麻醉后固定于手术台,沿腹中线剪开腹部3~4 cm,找到实验用肠段(十二指肠段自幽门1 cm处开始,空肠段自幽门15 cm处开始,回肠段自盲肠上行20 cm处开始,小肠自幽门1 cm 处开始至盲肠前端结束,结肠段从盲肠后端开始)取10 cm,在所取肠段两端剪一小口,插入塑料管,用丝线固定。生理盐水冲洗肠内容物,在肠端连接注射泵以0.5 mL·min-1 灌流药液,约30 min吸收稳定后,开始计时,用已知质量的小瓶在出口处每隔15 min收集1次,作为1个取样点,同时用下1个已知质量的小瓶替换,收集8次后完毕,计算收集前后小瓶质量称量差,测定收集液中药物浓度。实验结束后处死大鼠,用生理盐水冲洗,并剪下被灌流的肠段,测量其长度和内径。计算公式如下[15]。
Peff=-Qin·ln(Cout(cor)/Cin)2πrL
Ka=(1-Cout(cor)Cin)QinV
Fa=(1-Cout(cor)Cin)×100%
Cout(cor)=CoutQoutQin
Qout=Mout/Doutt
Qin为灌流液流速(mL·min-1);L为灌流肠段长度(cm);r为灌流肠段半径(cm);Cin为灌流液初始浓度(mg·L-1);Cout(cor)为经质量法校正后的灌流收集液浓度(mg·L-1);V=πr2L为灌流肠道体积(mL);Qout为通过灌流液密度测定的流出流速(mL·min-1);Cout为灌流收集液质量浓度(mg·L-1);Mout为灌流收集液质量(g),Dout为灌流收集液密度(g·mL-1),t为取样周期(min)。
吸收参数结果以±s表示,用统计分析软件SPSS 16.0处理数据,采用t检验分析,当P<0.05 时判定具有显著性差异。
3 结果
3.1 酚红药物浓度的测定
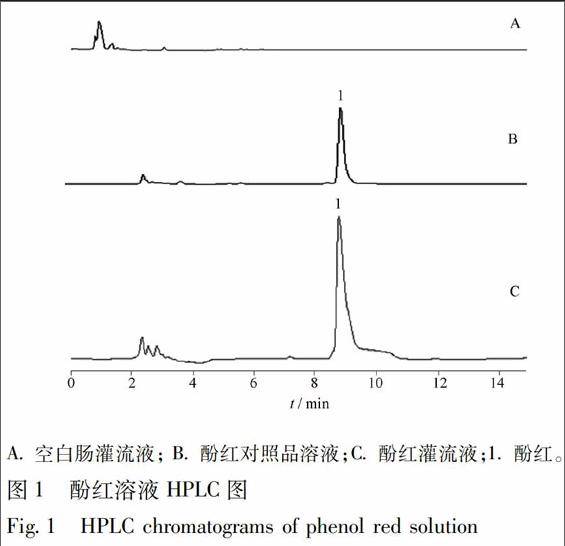
分别取空白灌流液、对照品溶液、含药灌流液进行液相检测,考察该色谱条件下空白灌流液对样品含量测定的干扰,结果见图1,在2.2.1.1色谱条件下,空白灌流液对样品含量测定无干扰,说明该方法的专属性良好,酚红的线性回归方程为Y=15 838X+3 367.9,R2=0.999 8,结果表明酚红在2.5~80 mg·L-1线性良好,测得的酚红的回收率为95.60%~103.4%,精密度、重复性和稳定性试验RSD均小于1%。
酚红肠吸收参数的计算结果(±s,n=3)有效渗透系数Peff为(1.09±0.62)×10-6 cm·s-1,吸收速率常数Ka为(1.19±6.7)×10-5 s-1,吸收分数Fa为(0.33±0.087)%,从结果可知,酚红为低渗透性药物,与文献报道相符[16-17],说明肠上皮细胞的紧密连接结构正常,且肠上皮的完整性保持良好。
3.2 地高辛浓度测定
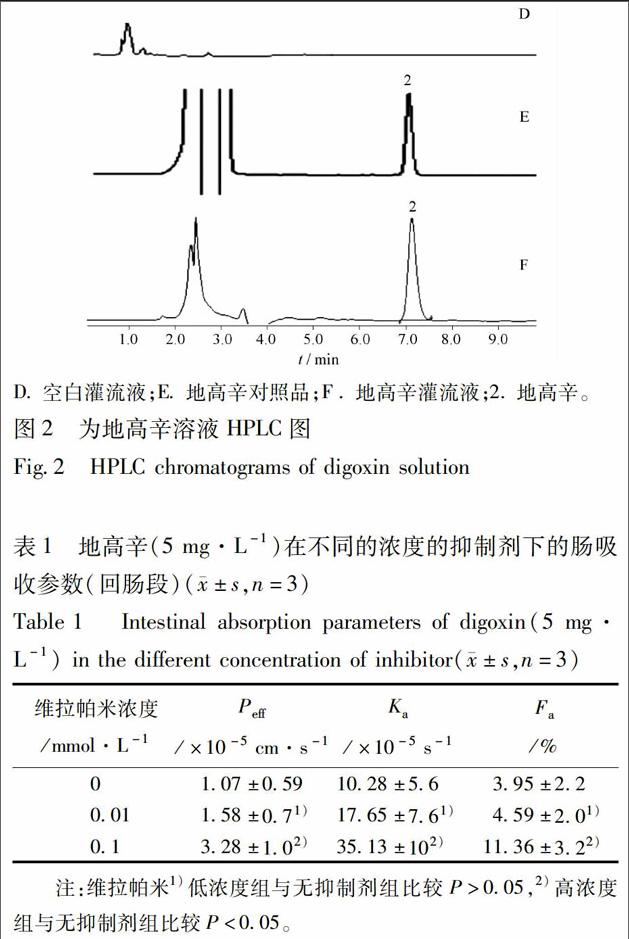
分别取空白灌流液、含药灌流液进行液相检测,考察该色谱条件下空白灌流液对样品含量测定的干扰,结果见图2,在1色谱条件下,空白灌流液对样品含量测定无干扰,说明该方法的专属性良好,地高辛的线性回归方程为Y=15 363X-2 499.7,r=0.999 1,地高辛在3.125~50 mg·L-1线性关系良好。高、中、低3个浓度的平均回收率95.56%~105.2%,精密度、重复性试验RSD均小于1%,稳定性试验小于2%。
当加入0.01 mmol·L-1维拉帕米时,对P-gp的抑制微弱,统计学显示P>0.05,差异不显著;给予0.1 mmol·L-1维拉帕米时,统计结果显示P<0.05,具有显著性差异,说明维拉帕米对P-gp的抑制程度与其浓度有关,不同浓度的维拉帕米对回肠段的P-gp抑制作用,随着浓度的增大而增强,见表1。实验结果表明P-gp在小肠的表达完整,模型具有可行性。
4 讨论
研究药物口服吸收的常用模型分为在体、离体和Caco-2细胞模型[18]。Caco-2细胞模型具有以下优点[19]:①研究药物从小肠吸收以往主要考虑的是从肠腔一侧(AP侧,apical side)摄入,实际上,药物从肠道进入循环系统,包括AP侧摄入和从肠壁一侧BL侧,(basolateral side)外排2個过程同时进行,用Caco-2细胞模型可以分别研究药物从AP侧的摄入,在细胞中的停留代谢及BL侧的外排等过程,因此对阐明药物在小肠中的吸收机制是十分有用的,动物实验则无法做到这些;②条件容易控制,数据重复性好;③与动物实验相比,更省时和经济。尽管Caco-2细胞模型在评价口服药物吸收方面有诸多的优点,但是也具有明显的不足[20]:①Caco-2是以结肠细胞系为基础分化形成的类似小肠组织,其紧密连接比小肠中实际存在的紧接连接更为紧密,这可能会导致基于该模型评价亲水性药物肠渗透时与体内实际吸收有一定差距;②虽然Caco-2细胞能表达活性转运蛋白系,但Caco-2 细胞中的转运蛋白种类和数量与机体小肠细胞仍有一定的差别;③机体肠细胞的黏液层是小肠吸收的主要屏障之一,但Caco-2细胞缺乏存在于小肠上皮细胞中的黏液层,这使得Caco-2 细胞的屏障特性与小肠上皮细胞也存在一定的差异。因而,Caco-2 细胞模型会导致对亲水性、通过细胞旁路和转运蛋白介导转运的化合物的肠渗透性预测能力变差。
外翻肠囊模型是研究药物肠吸收常用的离体模型[21],该模型的优点[22]是实验条件可控,减少了药物吸收的影响因素,获得的肠道吸收动力学参数更容易分析药物在肠道的吸收特征;该模型的缺点在于[23]:肠囊内液体停滞不动,这与机体小肠的蠕动状态的生理条件不同;另外,制备外翻肠囊时容易损伤肠粘膜,导致模型的肠壁细胞受损而影响模型的准确性。而在体灌流模型与体外实验相比最大的优点就是保持了小肠血管、淋巴和神经等主要生理系统的完好[9],且模型中保留了各种转运蛋白的完整性。该模型最接近机体内的状态,更符合正常的生理条件,从而能够更准确预测口服药物在体内的吸收情况。但在体灌流模型也存在一定的不足:需要的动物数量较大,成本高,不同动物组别之间具有一定的差异性。所以在进行药物肠渗透性研究时,考虑模型之间的差异很重要,有时需要几种模型方法联合应用,从而建立更好、更适宜的肠吸收预测模型,这样才能提高对药物的BCS分类判定的准确性。
P-gp 在小肠上皮细胞、肝胆管、肾小管及血脑屏障均有表达,它对经p-糖蛋白转运的药物的吸收、分布、排泄及药物相互作用均有明显影响[24],所以基于P-gp的相互作用是目前研究的较多因素之一。P-糖蛋白在大鼠小肠的分布为从高到低肠段依次增加[25],所以本实验选择P-gp相对含量较高的回肠作为渗透性研究的肠段。
P-gp具有ATP依赖性的药物外排泵的功能,能将细胞内的药物泵出细胞外使细胞内的药物浓度降低[26],因而肠道P-gp的功能活性和表达水平的变化可引起某些P-gp底物药物的口服生物利用度改变,常通过联合用药来抑制P-gp的外排作用是提高药物生物利用度的有效途径,基于此,采用P-gp 抑制剂与抗癌药物联合应用已经成为抗肿瘤治疗过程中对付多药耐药性的解决方案[27]。所以,建立基于P-糖蛋白的在体单向肠灌流模型,充分了解药物之间在P-gp环节可能产生的相互作用,不论对于合理地联合用药,避免药物毒性或不良反应,还是提高药物渗透性分类的准确性都至关重要。
[参考文献]
[1] Balimane P V, Chong S, Morrison R A. Current methodologies used for evaluation of intestinal permeability and absorption[J]. J Pharm Toxicol Methods, 2000, 44(1):301.
[2] Shah Vinod P, Amidon Gordon L. G.L. Amidon, H. Lennernas, V.P. Shah, and J.R. Crison. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability, Pharm Res 12, 413-420, 1995-Backstory of BCS[J]. AAPS J, 2014,doi: 10.1208/s12248-014-9620-9.
[3] Amidon G L, Lennernas H, Shah V P, et al. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability[J]. Pharm Res, 1995, 12 (3):413.
[4] Bene Leslie Z.The role of BCS (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classification system) in drug development[J]. J Pharm Sci, 2013, 102(1):34.
[5] 劉洋,隗丽,董玲,等. 多成分体系下中药生物药剂学分类系统的构建分析[J]. 中国中药杂志,2014,39(23):4479.
[6] 侯成波,汪国鹏,张强,等. 中药生物药剂学分类系统中多成分环境对葛根素溶解度的影响[J].中国中药杂志,2014,39(23):4494.
[7] 刘洋,王刚,董玲,等. 中药生物药剂学分类系统中多成分环境对葛根素渗透性的影响[J]. 中国中药杂志,2014,39(23):4505.
[8] 隗丽,朱美玲,董月柳,等. 盐酸小檗碱在黄连水煎液中的生物药剂学分类系统属性研究[J]. 中国中药杂志,2016,41(7):1192.
[9] Praveen V Balimane, Saeho Chong, Richard A Morrison, et al. Current methodologies used for evaluation of intestinal permeability and absorption[J]. J Pharm Toxicol Methods, 2000, 1(44):301.
[10] FDA. Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharm aceutics Classification System[EB/OL].[2016-11-14]. http://www.fda.gov/About FDA /Centers Offices/Office of Medical Products and Tobacco/CDER /ucm 128219.htm.
[11] Chen M L, Amidon G L, Benet L Z, et al. The BCS, BDDCS, and regulatory guidances[J]. Pharm Res, 2011, 28(7): 1774.
[12] Dezani T M, Dezani A B, da Silva Junior J B, et al. Single-pass intestinal perfusion (SPIP) and prediction of fraction absorbed and permeability in humans: a study with antiretroviral drugs[J]. Eur J Pharm Biopharm, 2016, 104: 131.
[13] Yang Y, Faustino P J, Volpe D A, et al. Biopharmaceutics classification of selected β-blockers: solubility and permeability class membership[J]. Mol Pharm, 2007, 4(4): 608.
[14] Chen M L, Yu L. The use of drug metabolism for prediction of intestinal permeability[J]. Mol Pharm, 2009, 6(1): 74.
[15] Li H, Dong L, Liu Y, et al. Comparison of two approaches of intestine absorption by puerain [J].Pharm Toxicol Methods, 2014, 70(1): 6.
[16] 崔升淼,赵春顺,何仲贵,等.大鼠肠管外翻模型对葛根素吸收机制的研究[J]. 时珍国医国药,2008,19(7):1715.
[17] 胡一桥,郑梁元,錢陈钦,等.离子型药物酚红的小肠吸收研究[J]. 中国药科大学学报,1996,27(6):355.
[18] 陈伟薇,李俊,宋珏.药物肠吸收的研究方法进展[J]. 安徽医药,2009,13(4):350.
[19] 卢智玲,冯怡,徐德生,等. Caco-2细胞模型在中药口服吸收及机制研究中的应用[J].中草药,2006,37(4):616.
[20] 高坤,孙进,何仲贵. Caco-2细胞模型在口服药物吸收研究中的应用[J]. 沈阳药科大学学报,2005,22 (6):469.
[21] 莫李立,王素军.口服药物吸收模型的研究进展[J].广东药学院学报,2011,27(1):104.
[22] 董宇,张英丰,杨庆,等.黄连提取物在大鼠肠外翻实验中的吸收研究[J]. 中国中药杂志,2008,33(9):1056.
[23] Amidon G E, Ho N F H, French A B,et al. Predicted absorption rates with simultaneous bulk fluid flow in the intestinal tract[J]. J Theor Biol, 1981,2(89):195.
[24] Engidawork E, Roberts J C, Hardmeier R, et al. Expression of the multidrug resistance P glycoprotein (P-gp) and multidrug resistance associated protein (MRP1) in down syndrome brains[M]//Lubec G.Protein Expression in Down Syndrome Brain. Vienna: Springer, 2001:35.
[25] Takara K, Ohnishi N, Horibe S, et al. Expression profiles of drug-metabolizing enzyme CYP3A and drug efflux transporter multidrug resistance 1 subfamily mRNAS in rat small intestine[J]. Drug Metab Dispos, 2003, 31(10):1235.
[26] 彭文兴,汪新亮,李焕德. 体内药物相互作用新点P-糖蛋白[J]. 中国临床药理学杂志,2001,17(5):386.
[27] 刘治军,傅得兴,孙春华,等. 体内药物相互作用研究进展[J]. 药物不良反应杂志,2006,8(1):33.
[责任编辑 孔晶晶]
猜你喜欢
小学阅读指南·教研版(2023年7期)2023-07-17
乐府新声(2021年4期)2022-01-15
基层中医药(2021年6期)2021-11-02
中华养生保健(2020年7期)2020-11-16
世界核地质科学(2018年2期)2018-07-05
生物医学工程学进展(2018年1期)2018-04-26
实用临床医学(2016年8期)2016-06-07
西藏科技(2015年9期)2015-09-26
中国当代医药(2015年30期)2015-03-01
中国当代医药(2015年10期)2015-03-01
