
在线红外光谱研究乌洛托品成盐机理
2017-05-07 07:04:25陈丽珍曹端林王建龙
含能材料 2017年5期
宋 亮, 陈丽珍, 曹端林, 王建龙
(中北大学化工与环境学院, 山西 太原 030051)
1 引 言
中红外光谱(mid infrared spectroscopy,MIR)的波长范围为2.5~25 μm,波数范围为4000~400 cm-1,反映的是分子中原子间的伸缩和变形振动运动。通过分析化合物的红外光谱可以获得化合物分子结构信息,根据光谱中吸收峰的位置和形状可以推断未知物的化学结构,根据特征吸收峰的相对强度可以测定混合物中各组分的含量。
含能化合物的合成很少是能一步完成的基元反应,大多都要经过多步反应和复杂的过程。要了解一个反应的机理,就必须了解整个反应中复杂变化的信息。有时反应过程非常迅速,用离线分析法很难捕获过程信息。因此,近几年发展的在线红外光谱技术[1-5],利用探头的衰减全反射原理[6-7],实时跟踪化学反应全过程,可以提供整个反应过程的信息,根据在线谱图中特征吸收峰的变化捕捉反应过程中出现的中间体或副产物,可推测出合理的反应机理。
乌洛托品是工业上生产黑索今(RDX)的重要原材料。乌洛托品在硝酸中的硝解反应比较复杂,除了生成RDX外,还有一系列的副反应产物。几十年来国内外学者已做了不少有关RDX制备过程中硝解反应机理研究[8-10],见解不一,并且存在一定的局限性。普遍认为HA在浓硝酸中作用下,首先形成HADN,然后在不同的位置断键,进一步硝解,生成RDX、HMX、1,7-二硝酸酯基-2,4,6-三硝基-2,4,6-三氮杂庚烷(ATX)等产物。但是,郁波[11]用高效液相色谱分析法跟踪了该硝解反应过程,得出结论,HA反应过程中首先生成HAMN,而不是HADN。为了进一步了解HA的成盐过程,明确成盐机理,本研究通过在线红外技术监测HA与NA生成HAMN和HADN过程,对反应过程的主要官能团特征吸收峰进行跟踪分析,并利用计算化学方法研究乌洛托品与硝酸之间的相互作用,进而推测可能的反应机理,为研究乌洛托品硝解机理提供理论支持。
2 实验
2.1 仪器和试剂
VERTEX80型在线红外光谱仪(Bruker Optics,Ettlingen,Germany),ATR探头(IN350-T,Germany)。
乌洛托品,分析纯,常州市武进化工厂; 硝酸,98%,天津市化学试剂研究所; 乙醇,分析纯,天津市北辰方正试剂厂; 去离子水,实验室自制。
2.2 实验步骤
在线红外参数设置: 分辨率4 cm-1,扫描范围4000~650 cm-1,测量背景单通道扫描次数为32; 每间隔2 s对样品进行扫描,采集一张红外谱图。
(1)在线红外监测乌洛托品一硝酸盐的生成过程: 在室温条件下,将15 g乌洛托品溶于30 mL水中,配置成乌洛托品水溶液,然后放在冰水浴中冷却到5 ℃以下。扫描乌洛托品的水溶液作为背景,缓慢滴加6.75 mL 70%硝酸,加料时保持温度低于 5 ℃,加料完毕后继续搅拌5 min,利用ATR探头在线监测HAMN的生成过程。然后将反应液进行减压蒸发,使溶液浓缩到原溶剂的1/3,降温至室温。加入酒精使晶体析出。过滤,用酒精洗涤,得到HAMN。
(2)在线红外监测乌洛托品二硝酸盐的生成过程: 在室温条件下,将20 g乌洛托品溶于35 mL水中,配置成乌洛托品水溶液,然后放在冰水浴中冷却到10~15 ℃,扫描乌洛托品的水溶液作为背景,缓慢滴加22 mL 70%硝酸,保持温度不超过15 ℃,加料完毕后在5 ℃继续搅拌15 min,利用ATR探头在线监测HADN的生成过程。直接将HADN过滤出来,再用乙醇洗涤。
3 结果与讨论
3.1 原料的红外谱图
分别采集HA固体、H2O/HA混合溶液以及硝酸溶液的红外谱图,其中H2O/HA混合溶液的红外谱图是在KBr压片上滴加2.2(2)中浓度的混合溶液测得的,硝酸溶液的红外谱图是以空气为扫描背景把ATR探头放入98%发烟硝酸溶液得到吸光度谱图,如图1所示。由图1a可发现,HA中C—N键的对称和反对称伸缩振动峰[10-12]分别位于1006 cm-1和1238 cm-1,弯曲振动频率为672 cm-1和812 cm-1; H2O/HA混合溶液中,HA中的C—N键的对称和反对称伸缩振动频率分别位于1011 cm-1和1238 cm-1,弯曲振动频率为672 cm-1和812 cm-1; 水中—OH键的伸缩振动峰位于3436 cm-1,变角振动吸收峰位于1646 cm-1。由图1b可知,硝酸溶液中O—NO3的对称和反对称伸缩振动频率为1265 cm-1和1654 cm-1,由于硝酸溶液中存在大量的氢键,—OH的呈现为宽谱带,其伸缩振动峰位于3277 cm-1。
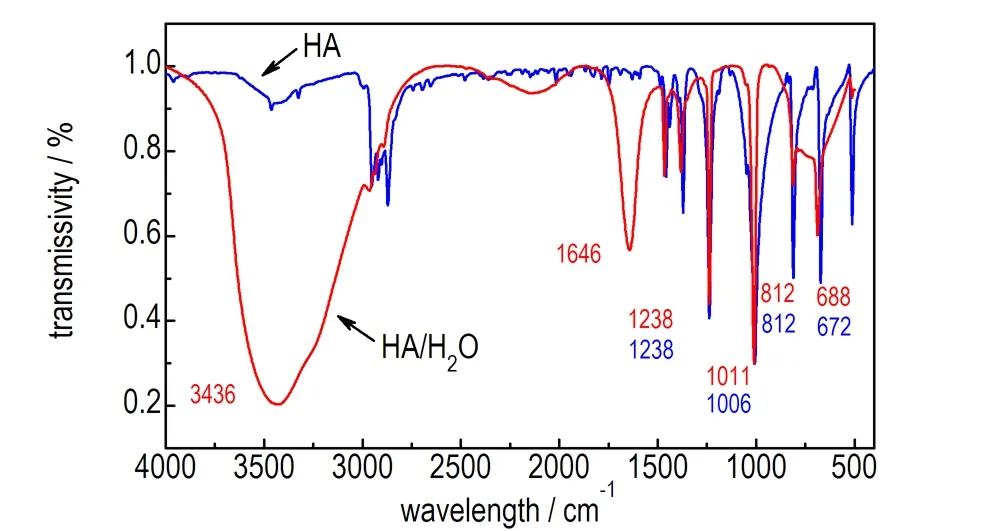
a. HA and H2O/HA
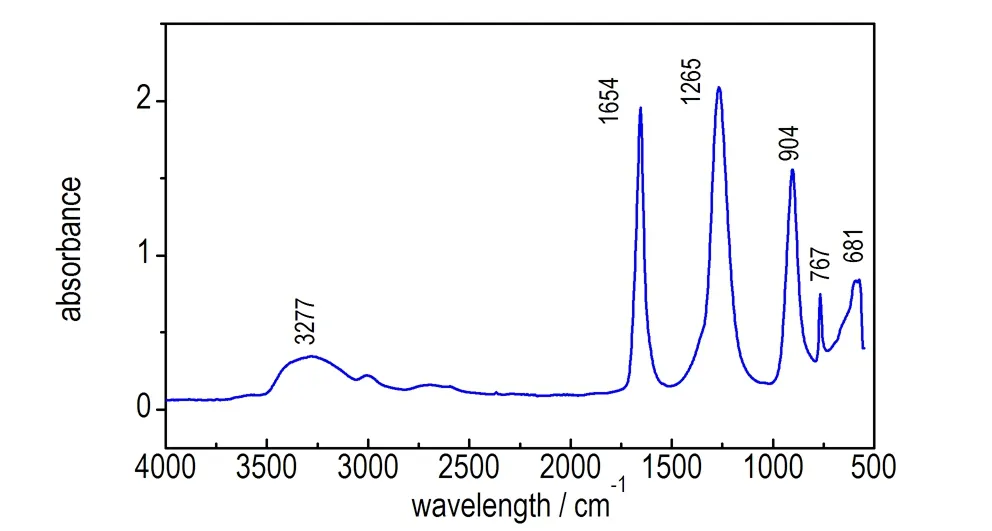
b. HNO3
图1 HA与H2O/HA红外谱图以及HNO3溶液红外吸光度谱图
Fig.1 Infrared spectra of HA and H2O/HA and infrared absorbance spectrum of HNO3solution
3.2 反应过程中红外光谱的变化
图2所示为反应过程中的三维谱图,其中图2a为HAMN合成过程的三维红外谱图,图2b为HADN合成过程的三维红外谱图。可以看出,随着反应进行,体系中的红外谱图均发生了较大的变化,下面通过分别讨论HAMN和HADN生成过程中红外光谱图的变化来推断HA和HNO3的相互作用。
3.2.1 HAMN生成过程的红外光谱分析
图3是以H2O/HA混合溶液为扫描背景,波数范围选择为1500~500 cm-1,采集HAMN生成过程中的红外谱图,取整百数秒观察反应过程中的谱图的变化情况。
从图3中可以看到在1002 cm-1和1236 cm-1处出现了颠倒的峰(扣除背景所致),通过与图1中HA的比较,确定为C—N键的对称和反对称伸缩振动吸收峰[13],说明在反应过程中HA在不断消耗。随着反应的进行,在979,1024,1219,1259 cm-1(与硝酸中的O—NO3的1265 cm-1峰重合,导致出现较强的吸收峰)处出现吸收峰。其中979 cm-1处和1024 cm-1处的吸收峰为C—N键的对称伸缩振动吸收峰,1219 cm-1和1259 cm-1处的吸收峰为C—N键的反对称伸缩振动吸收峰。HA中的N原子与硝酸中的H原子结合时,引起化学键的力常数发生变化,在紧挨着吸收频率为1002 cm-1和1236 cm-1处,产生4个吸收峰,可以判定为两种不同的C—N键,一种C—N的对称和反对称伸缩振动吸收峰在979 cm-1和1219 cm-1处; 另一种C—N的对称和反对称伸缩振动吸收峰在1024 cm-1和1259 cm-1处。
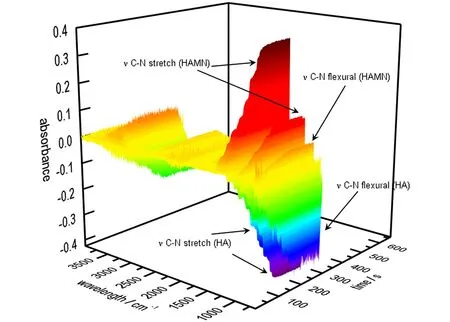
a. HAMN
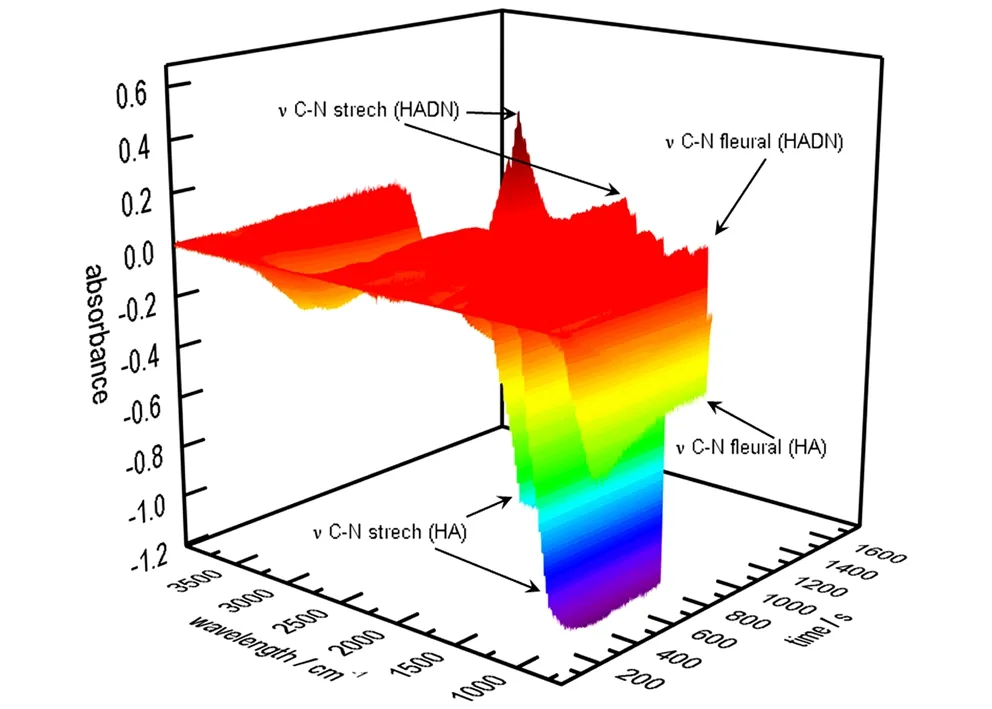
b. HADN
图2 在线监测HAMN和HADN的生成过程
Fig.2 On-line monitoring of the generative process of HAMN and HAND
在690 cm-1和806 cm-1处也出现颠倒峰,这是由于HA消耗造成C—N键弯曲振动的数量减少,从而引起吸光度的降低。同样,在690 cm-1和806 cm-1左右两边分别出现2个对应的吸收峰,与C—N的伸缩振动吸收峰呈现一致性。
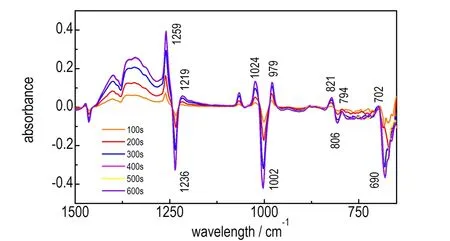
图3 HAMN生成过程中每100 s的红外光谱图
Fig.3 Infrared spectra of every 100 s during HAMN generation
3.2.2 HADN生成过程的红外光谱分析
在生成HADN的过程中,观测到有HADN析出现象。由图4中的200 s处的红外光谱发现,在1234,1002,807,681 cm-1处有倒峰出现,并在倒峰位置两边有较强的吸收峰,通过对比图3中HAMN生成过程中红外谱图里特征峰的位置和形状,可以判断在生成HADN过程中优先生成HAMN。在400~1600 s的范围,1234,1002,807,681 cm-1处倒峰的峰形几乎重合,吸光度值未发生太大变化,说明乌洛托品水溶液中的HA成分在400 s时已经基本消耗完全,而倒峰两边产生的HAMN中C—N吸收峰的强度却在变弱,1000 s处HAMN的C—N吸收峰已经消失。在整个反应过程中,并没有明显检测到HADN的特征吸收峰,这可能是HADN在溶液中的溶解度不大,生成后很快就析出,导致没有明显的检测到HADN的吸收峰。
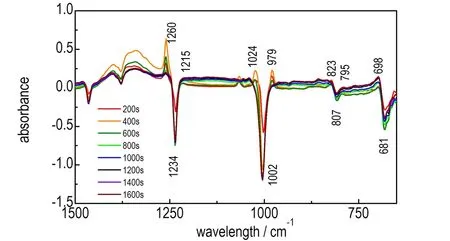
图4 HADN生成过程中每200 s的红外光谱图
Fig.4 The infrared spectra of every 200 s of during HADN generation
在1267 cm-1处的O—NO2对称伸缩振动吸收峰与HAMN的吸收峰重合,只能通过1633 cm-1处O—NO2的反对称伸缩振动吸收峰来判断硝酸在溶液体系的变化情况[14],如图5所示,在386 s 内,随着滴加硝酸溶液,硝酸的吸光度并没有大幅度的上升,这是因为HA的成盐属于酸碱中和反应,反应十分迅速。在HADN的析出过程中,停止滴加70%硝酸溶液,由于HADN的生成消耗了硝酸分子,在386~526 s之间,O—NO2吸光度值略微下降。然而,在526~874 s监测到的硝酸吸收峰并没有呈现继续下降的趋势,反而呈现出小幅度的上升。可能的解释为: 当HAMN和周围的硝酸能形成由弱相互作用的超分子体系,当硝酸增加到一定量时,生成HADN,并析出。同时,释放了硝酸分子,供给HAMN持续的生成新的HADN使用。874 s后,硝酸的吸光度基本不发生变化,反应基本完成。
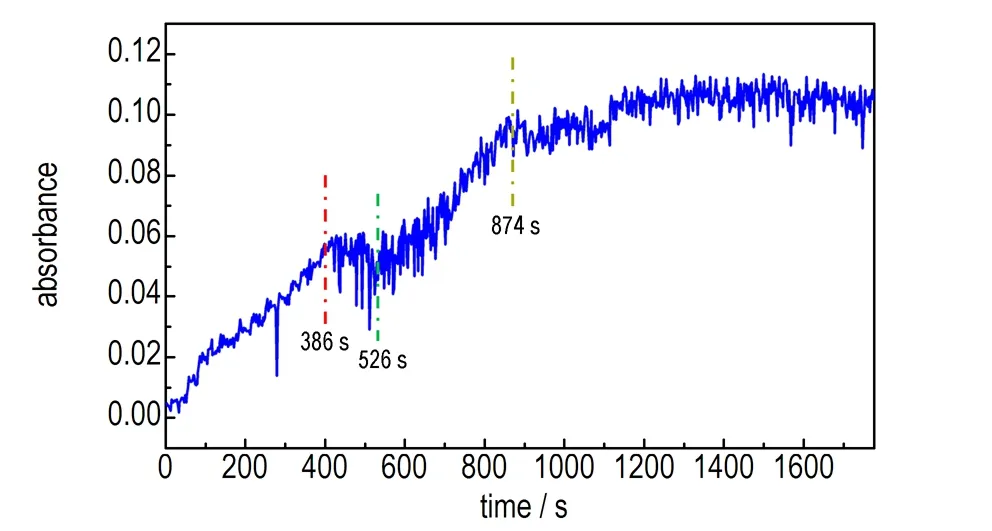
图5 O—NO2的反对称伸缩振动峰吸光度随时间的变化图
Fig.5 Change in the absorbance of O—NO2antisymmetric stretching vibration peak with time
3.3 产物的红外谱图
图6是将试验中所得到纯的HAMN和HADN经KBr压片测得的红外谱图。由于分子在固相晶格中排列非常有序,吸收峰比较尖锐,从图6中可以看到峰的数目明显增多。其中,纯的HAMN的C—N键的伸缩振动吸收峰分别为1253,1203,1009,977 cm-1,固态压片HAMN中C—N键发生红移,向低波数移动,相比于生成HAMN过程中溶液体系C—N键的伸缩振动频率依次减小了6,16,15,2 cm-1。可能的解释为固态HAMN分子间距离缩短,相互之间的吸引力加大,基团的振动波数低于液态。
固体HADN的红外谱图在1260~975 cm-1处C—N键的反对称和对称伸缩振动区域内变化相对复杂。主要是由于HADN比HAMN多出一个NA分子,通过静电诱导作用,引起HA环上电子分布变化较大。
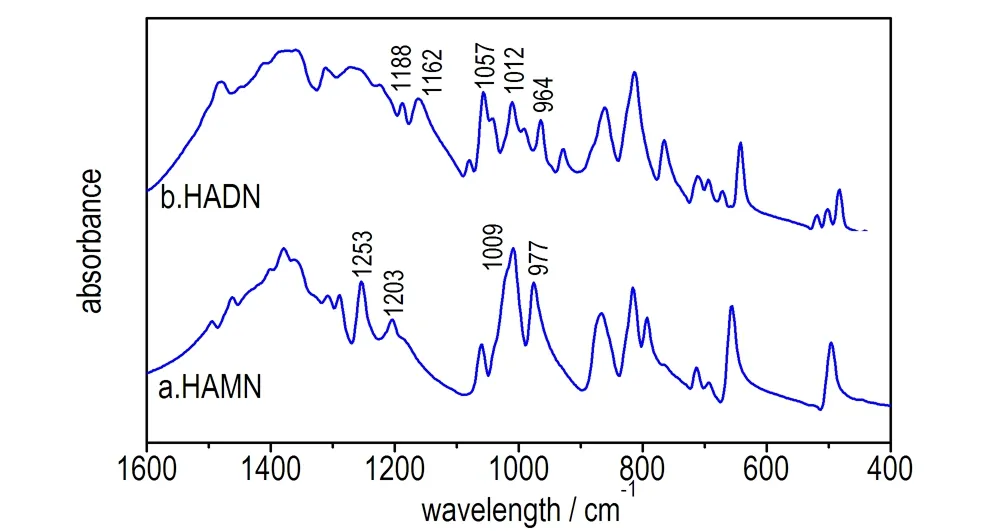
图6 HAMN和HADN的红外谱图
Fig.6 IR spectra of HAMN and HADN
4 HA硝酸盐的量子化学计算
4.1 计算方法
研究表明,密度泛函理论(DFT)是含能材料分子结构优化的一种有效理论计算方法。因此,本研究选用B3LYP方法在6-31G基组水平上对HA单体及其复合物进行结构优化,得到了势能面的最低点,即为能量最低分子最稳定的结构。在优化的结构上,用B3LYP/6-31G计算了分子间的结合能。所有计算使用Gaussian03软件包进行,收敛精度为程序内定值。
4.2 HA与1~4个NA分子的作用
综上所述,HA在NA体系下很容易生成HAMN,但HADN不是伴随着HAMN的生成而生成的,当硝酸浓度达到5.97 mol·L-1时才会生成HADN。运用计算化学方法对HAMN和HADN的生成机理做更进一步研究。
如图7所示,在HA中每个氮原子都处于四面体的顶端,在分子中彼此都是等价的; 每个碳原子都处于八面体的顶端,它们在分子中也彼此都是等价的。HA中的所有C—N键键长为1.494 Å,C—H键键长为1.094 Å; HNO3中O(2)—H(1)键键长为0.975 Å,O(3)—N(1)键键长为1.408 Å,N(1)—O(2)和N(1)—O(1)键长分别为1.218 Å、1.203 Å。
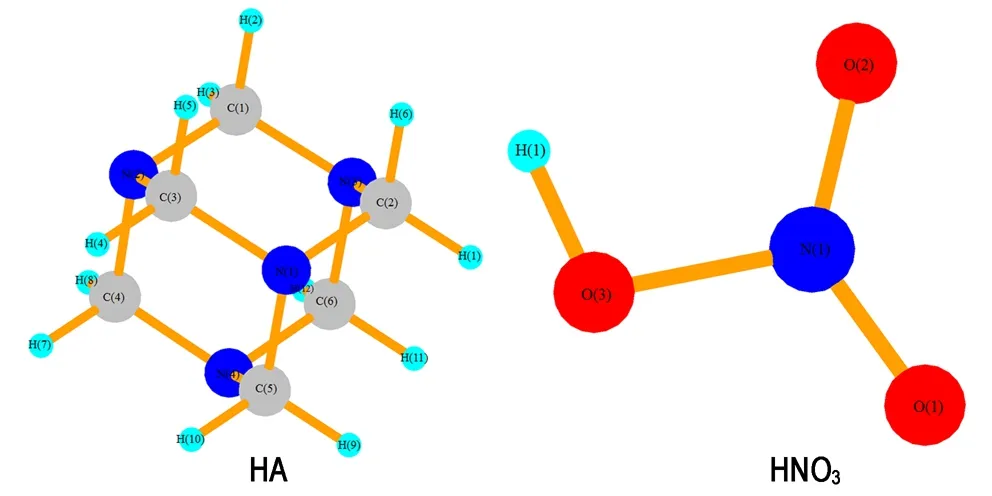
图7 HA和HNO3的结构优化结构
Fig.7 The optimized structure of HA and HNO3
在HA成盐过程中,反应液中包含了数目巨大的原子和分子,要完全定量地去描述这样的体系是不可能的,只能通过小的复制体系去模拟宏大体系的行为。运用Gaussian 03软件包对HA和1~4个HNO3分子进行了结构优化,进而讨论该体系的微观构型和性质。图8给出了HA·HNO3、HA·2HNO3、HAMN·3HNO3和HA·4HNO3优化后的空间构型。表1给出了HA与1~4个HNO3分子相互作用时N—H…O的键长变化情况。图8a中的结构为HAMN,HNO3分子中的H(13)已经与HA分子中的N(2)形成共价键(1.115 Å),而硝酸根与N(2)—H(13)在同一方向上,有氢键的特征,键长为1.468 Å。冯晓琴[15]已经对HA与两个NA的分子体系做过研究,她是以NA中的羟基氢作为电子受体,HA中的胺基氮作为电子供体对其结构进行优化。考虑到NA分子除了与HA中的氨基氮形成氢键外,还有可能与HAMN中的硝酸根形成氢键。因此,本文将NA中的氢为电子受体,硝酸根中的氧为电子供体进行研究,图8b和图8c展示NA分子与HAMN中的硝酸根形成强氢键。

表2展示了HA分子在不同体系中优化后的键长参数。可以发现与形成N—H键相邻的C—N键距离较长(1.5 Å左右),均大于单体HA中C—N键的距离(1.494 Å); 紧挨着H—N—C键的C—H键键长缩短(1.46 Å左右),剩下的C—N键组成的六元环中的C—N键键长在1.49 Å左右,与单体HA中的C—N键键长保持一致,说明N—H键的形成对其影响较小。根据键长的变化说明在HAMN生成的过程中,HA中的C—N键的力常数既有增大的部分又有减小的部分,对应着在每个HA特征峰位置两边产生两个不同的C—N键的特征吸收峰。从表2中还可以发现随着NA的增多,与N—H相邻的C—N键的键长逐渐增大,由1.54 Å增加到1.56 Å,根据最弱键断裂原则可知,在化学反应中,与形成N—H键相邻的三个C—N键最容易断裂。
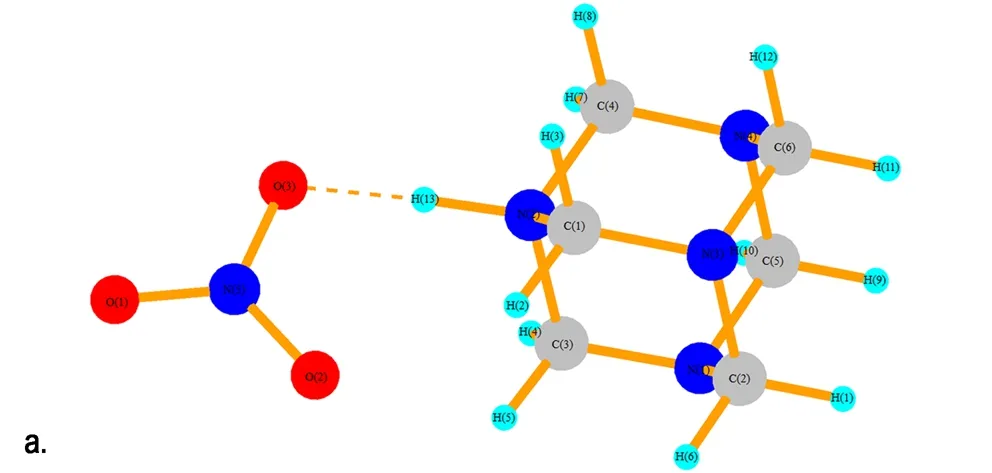
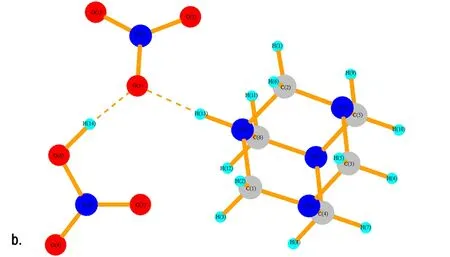
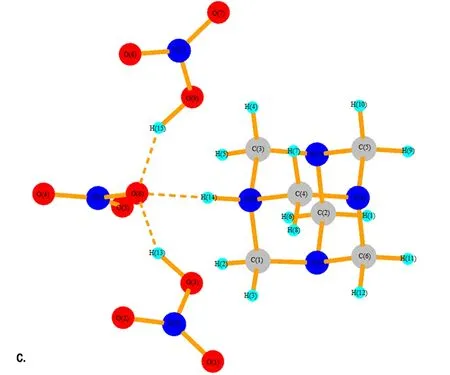
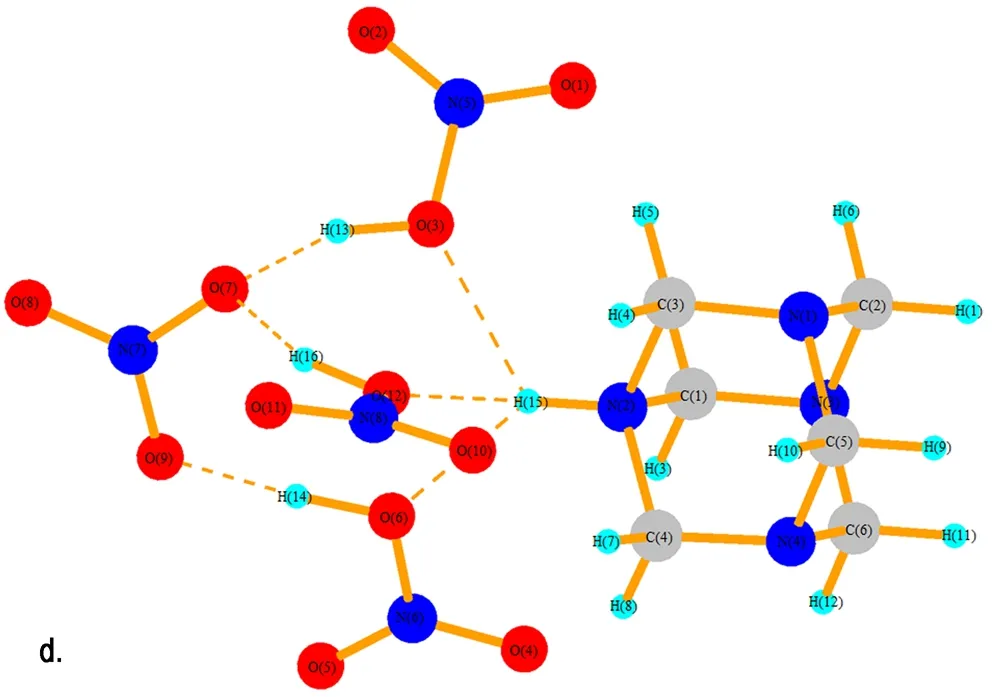
图8 HA·(1~4)HNO3的优化结构
Fig.8 The optimized structure of HA·(1-4)HNO3
表1 优化后主要的N—H键和O—H键的键长
Table 1 The main bond length of C—H bond and O—H bond after optimizing

molecularstructurebondlength/Åmolecularstructurebondlength/ÅHA·HNO3N(2)—H(13)1.115HA·3HNO3N(2)—H(14)1.046O(3)—H(13)1.468O(6)—H(14)1.771HA·2HNO3N(3)—H(13)1.065HA·4HNO3N(2)—H(15)1.028O(3)—H(13)1.629--
表2 优化后HA分子的主要键长
Table 2 The main bond length of HA molecule after optimizing
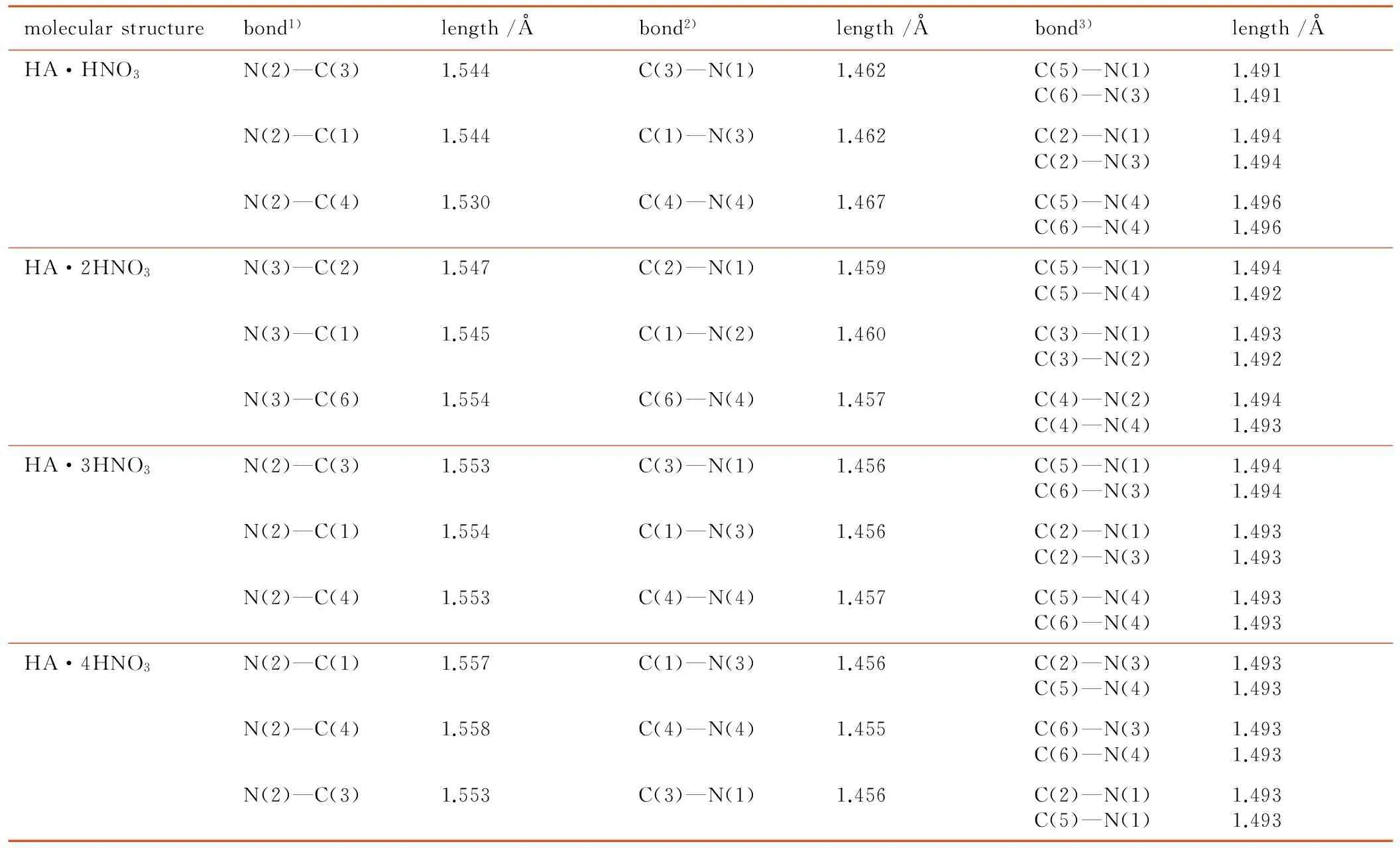
molecularstructurebond1)length/Åbond2)length/Åbond3)length/ÅHA·HNO3N(2)—C(3)1.544C(3)—N(1)1.462C(5)—N(1)C(6)—N(3)1.4911.491N(2)—C(1)1.544C(1)—N(3)1.462C(2)—N(1)C(2)—N(3)1.4941.494N(2)—C(4)1.530C(4)—N(4)1.467C(5)—N(4)C(6)—N(4)1.4961.496HA·2HNO3N(3)—C(2)1.547C(2)—N(1)1.459C(5)—N(1)C(5)—N(4)1.4941.492N(3)—C(1)1.545C(1)—N(2)1.460C(3)—N(1)C(3)—N(2)1.4931.492N(3)—C(6)1.554C(6)—N(4)1.457C(4)—N(2)C(4)—N(4)1.4941.493HA·3HNO3N(2)—C(3)1.553C(3)—N(1)1.456C(5)—N(1)C(6)—N(3)1.4941.494N(2)—C(1)1.554C(1)—N(3)1.456C(2)—N(1)C(2)—N(3)1.4931.493N(2)—C(4)1.553C(4)—N(4)1.457C(5)—N(4)C(6)—N(4)1.4931.493HA·4HNO3N(2)—C(1)1.557C(1)—N(3)1.456C(2)—N(3)C(5)—N(4)1.4931.493N(2)—C(4)1.558C(4)—N(4)1.455C(6)—N(3)C(6)—N(4)1.4931.493N(2)—C(3)1.553C(3)—N(1)1.456C(2)—N(1)C(5)—N(1)1.4931.493
Note: 1)The C—N bond adjacent to the N—H bond. 2)The C—N bond adjacent to the H—N—C bond. 3)The remaining C—N bond on hexatomic ring.
Ebinding energy=Efragment 1+Efragment 2-Etotal
(1)
在B3LYP/6-31G水平上分别对HA(片段1)和1~4个NA分子(片段2)的结合能(式(1))进行计算。计算结果见表3,结合能为正值,属于放热反应。随着NA分子数的增多,放出的热量逐渐增大,复合体越稳定。当NA分子数达到4时,结合能的值比HA分子与一个NA分子结合所释放能量的2倍还多。由此可知,HAMN极容易吸附NA分子,并以氢键结合。
4.3 HA与1~4个H2O分子的作用
在反应液中,除了有HNO3分子外,还存在大量的H2O分子。同样,运用Gaussian 03软件包在B3LYP/6-31G水平上分别对HA和1~4个H2O分子进行了结构优化。计算结果如图9所示,H2O与HA分子体系主要以复合体的形式存在,其中结构a~d中氢键(N…H键)的距离依次为1.894 Å、1.744 Å、1.839 Å和1.871 Å,其中b结构中N…H键的缩小是由于H2O中O(2)与HA中的H(4)和H(5)形成弱氢键作用,导致两个H2O分子整体向HA方向移动。在c结构和d结构中新添加的H2O分子更倾向于与体系中H2O分子中的氧原子形成氢键。由表5可见,HA与H2O的结合能的值远小于HA与NA的结合能的值。因此,在溶液中的NA分子与HA分子结合能力更大。
表3 HA·(1~4)HNO3总能量与校正结合能的数据参数
Table 3 Data parameters for the total energy and corrected binding energy of HA·(1-4) HNO3
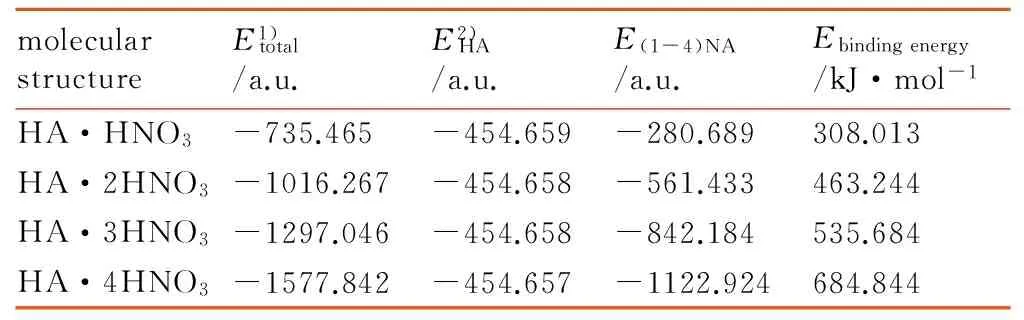
molecularstructureE1)total/a.u.E2)HA/a.u.E(1-4)NA/a.u.Ebindingenergy/kJ·mol-1HA·HNO3-735.465-454.659-280.689308.013HA·2HNO3-1016.267-454.658-561.433463.244HA·3HNO3-1297.046-454.658-842.184535.684HA·4HNO3-1577.842-454.657-1122.924684.844
Note: 1)The uncorrected total energy; 2)The corrected total energy of fragments from Basis Set Superposition Error (BSSE) .
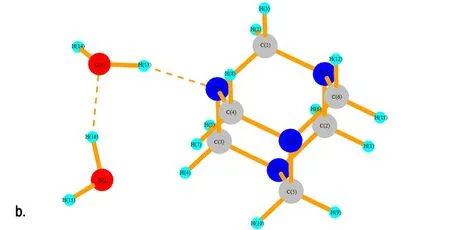
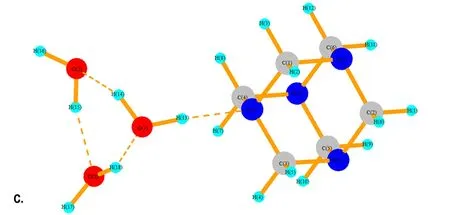
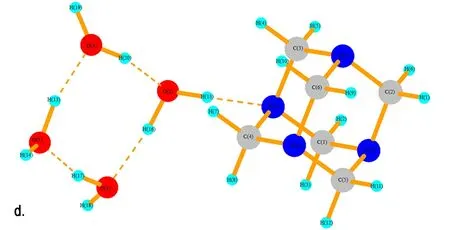
图9 HA·(1~4)H2O的优化结构
Fig.9 The optimized structure of HA·(1-4)H2O
表4 HA·(1~4)H2O总能量与校正结合能的数据参数
Table 4 Data parameters for the total energy and corrected binding energy of HA·(1-4) H2O
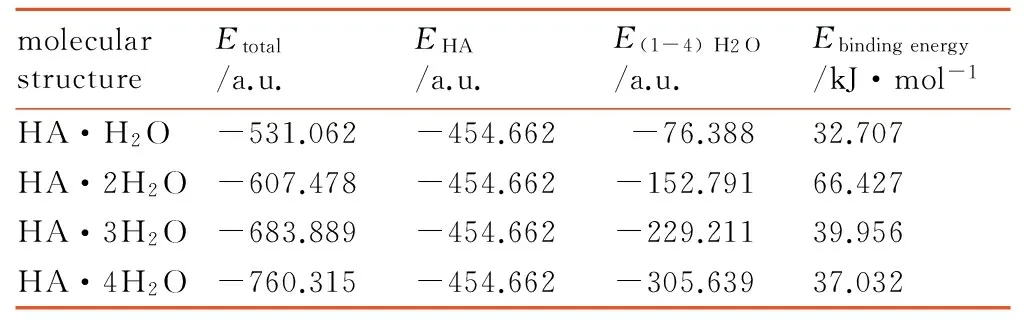
molecularstructureEtotal/a.u.EHA/a.u.E(1-4)H2O/a.u.Ebindingenergy/kJ·mol-1HA·H2O-531.062-454.662-76.38832.707HA·2H2O-607.478-454.662-152.79166.427HA·3H2O-683.889-454.662-229.21139.956HA·4H2O-760.315-454.662-305.63937.032
4.4 HADN的计算化学研究及HADN断键的探索
在计算HA和两个NA分子作用时,只有一个硝酸中的H与HA成键,第二个硝酸中的H与HA以复合体的形式存在。考虑到HA可能在大量硝酸分子的相互作用下才能形成与两个H成共价键的HADN分子。在B3LYP/6-31G水平上对HA和多个NA分子体系进行结构优化。当NA分子数达到13时如图10所示。图10a为优化过程中的一帧,位置1的氢已经与氮原子形成共价键,键长为1.069 Å; 与硝酸根中氧原子形成氢键,键长1.641 Å。同样,在位置2的氢与氮原子形成的键也为共价键,键长为1.045 Å; 与硝酸分子中的氧原子形成氢键,键长为1.761 Å,位置3的硝酸根游离出去,与其它NA分子形成氢键作用。说明在生成HADN的形成过程中,需要足够多的NA分子,HA才能够和两个H形成化学键。图10b为优化收敛后的结果,位置4和5的N—H键的键长分别为1.048,1.041 Å。位置6和7为游离出去的硝酸根。
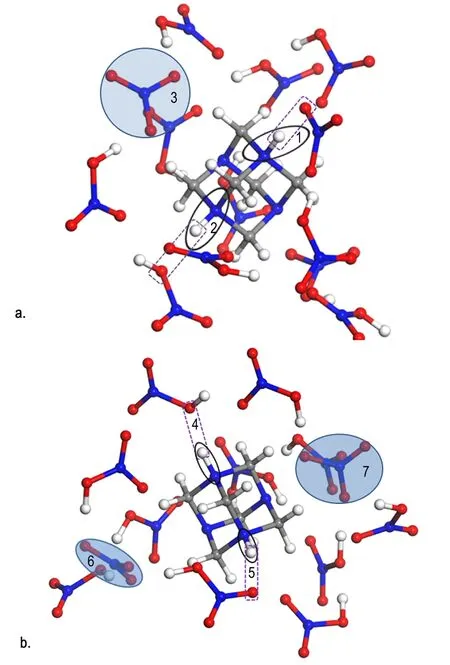
图10 HA·13H2O的优化结构
Fig.10 The optimized structure of HA·13H2O
由于体系复杂,从头算对服务器性能要求较高,并且耗时长,因此,未对能量和反应动力学做进一步计算,仅通过键长来判断键的强弱。图11是提取图10b中的结构片段,收敛后的结构参数见表5,由于氢原子是斥电子基团,与N—H相邻的C—N键键长都被拉长,但N(1)—C(3)和N(2)—C(3)的键长短于其它与N(1)和N(2)形成的C—N键键长。根据最弱键断裂原则可知,如果断两个键存在四种的可能方式。如图11将其归为两类,一种为对位断键,将留下一个带两个亚甲基的八圆环带结构,该结构可能进一步反应生成HMX。另一种为邻位断键,变成一个主体为六圆环的结构,可以进一步硝解成RDX。
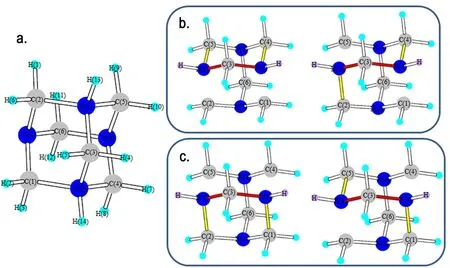
图11 HADN的断键位置
Fig.11 The bond-breaking position of HADN
表5 优化后HA分子的主要键长
Table 5 The main bond length of HA molecule after optimizing
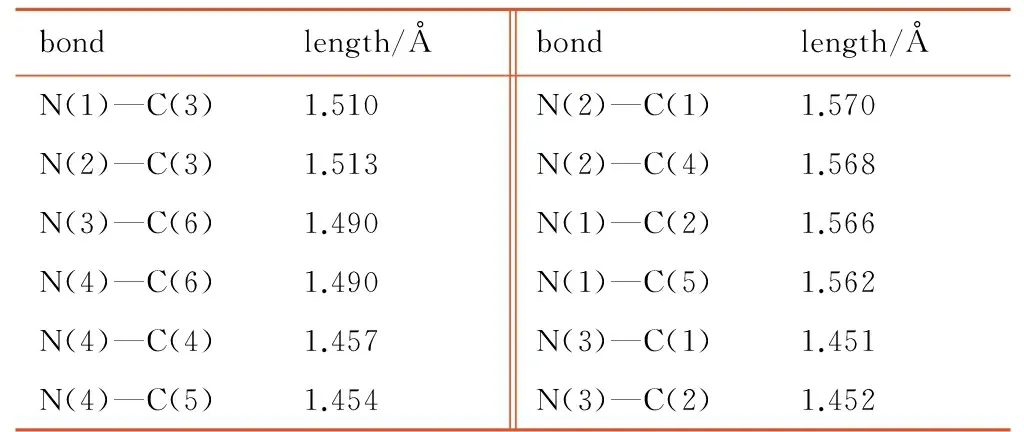
bondlength/Åbondlength/ÅN(1)—C(3)1.510N(2)—C(1)1.570N(2)—C(3)1.513N(2)—C(4)1.568N(3)—C(6)1.490N(1)—C(2)1.566N(4)—C(6)1.490N(1)—C(5)1.562N(4)—C(4)1.457N(3)—C(1)1.451N(4)—C(5)1.454N(3)—C(2)1.452
4 结 论
(1) 利用在线红外光谱分析了乌洛托品的成盐过程,对采集的红外谱图进行分析,结果表明HNO3与HA生成HAMN的反应很快,而HAMN到HADN的反应相对较慢。当硝酸达到5.97 mol·L-1时,才有HADN生成。
(2) 当有HADN析出时,不滴加硝酸,依然有HADN生成并析出,且溶液中硝酸的量呈现缓慢上升趋势。与量子化学计算结果结合分析,当生成HAMN过程中,吸附了周围的硝酸分子,与之形成强氢键作用,当有HADN生成时,释放硝酸分子到溶液中,供给HAMN分子继续反应生成新的HADN分子。由于与HAMN分子缔合的HNO3分子个数大于生成HADN分子所需HNO3分子的个数,致使溶液中监测的硝酸特征吸收峰吸光度不断增大。
(3) 通过在线红外光谱中的HAMN谱图可以看到,在1002,1236,690,806 cm-1处出现颠倒的峰,这些峰为HA中C—N键的特征吸收峰。在这些特征峰两边均出现新的特征吸收峰,这是由于HAMN的生成,引起化学键的力常数发生改变。通过量化计算结果可知,与N—H键相邻的C—N键伸长,紧挨着H—N—C键的C—H键的键长缩短,而最后的六元环上的C—H键变化不大。
(4) 对HA和多个NA分子体系进行优化,发现HA能与两个H原子形成共价键,但分子体系相对复杂,有待于进一步研究。
参考文献:
[1] Lumpi D, Wagner C, Schöpf M, et al. Fibre optic ATR-IR spectroscopy at cryogenic temperatures: in-line reaction monitoring on organolithium compounds[J].ChemicalCommunications, 2012, 48(18): 2451-3.
[2] Fernàndez-Francos X, Kazarian S G, Ramis X, et al. Simultaneous monitoring of curing shrinkage and degree of cure of thermosets by attenuated total reflection Fourier transform infrared (ATR FT-IR) spectroscopy[J].AppliedSpectroscopy, 2013, 67(12): 1427.
[3] Gabrienko A A, Morozov E V, Subramani V, et al. Chemical visualization of asphaltenes aggregation processes studied in situ with ATR-FTIR spectroscopic imaging and NMR imaging[J].JournalofPhysicalChemistryC, 2015: 150123100456000.
[4] Kunimatsu K, Miyatake K, Deki S, et al. Analysis of the gold/polymer electrolyte membrane interface by polarization modulated ATR-FTIR spectroscopy[J].JournalofPhysicalChemistryC, 2015, 119(29): 16754-16761.
[5] Roberts J E, Zeng G, Maron M K, et al. Measuring heterogeneous reaction rates with ATR-FTIR spectroscopy to evaluate chemical fates in an atmospheric environment: A physical chemistry and environmental chemistry laboratory experiment[J].JournalofChemicalEducation, 2016, 93(4): 733-737.
[6]Bellamy M K. Using FTIR-ATR spectroscopy to teach the internal standard method[J].JournalofChemicalEducation, 2010, 87(12): 1399-1401.
[7] And Y J, Zhu X Y. FTIR spectroscopy of buried interfaces in molecular junctions[J].JournaloftheAmericanChemicalSociety, 2004, 126(41): 13224-13225.
[8] Greenfield M, Guo Y Q, Bernstein E R. Ultrafast photodissociation dynamics of HMX and RDX from their excited electronic states via femtosecond laser pump-probe techniques[J].ChemicalPhysicsLetters, 2006, 430(4-6):277-281.
[9] Radhakrishnan S, Talawar M B, Venugopalan S, et al. Synthesis, characterization and thermolysis studies on 3,7-dinitro-1,3,5,7-tetraazabicyclo[3,3,1]nonane (DPT): A key precursor in the synthesis of most powerful benchmark energetic materials (RDX/HMX) of today[J].JournalofHazardousMaterials, 2008, 152(3): 1317.
[10] 方世杰, 陈驹, 王绍芳, 等. 用核磁共振法研究六次甲基四胺的硝解反应—次甲基二硝胺的分解产物和生成黑索今的中间体[J]. 火炸药学报, 1992(3): 1-4.
FANG Shi-jie, CHEN Ju, WANG Shao-fang, et al. Nuclear magnetic resonance spectroscopy of hexamethy lenetetramine nitrate solution reaction—methylene nitramine decomposition products and generation of RDX intermediates[J].ChineseJournalofExplosives&Propellants, 1992(3): 1-4.
[11] 郁波. 酸性离子液体催化下直接法合成黑索金及硝解机理研究[D]. 南京: 南京理工大学, 2009.
YU Bo. Study on the direct synthesis of RDX and nitrification mechanism catalyzed by acidic ionic liquid[D]. Nanjing: Nanjing University of Science and Technology, 2009.
[12] 苏克曼, 潘铁英, 张玉兰, 等. 波谱解析法[M]. 上海: 华东理工大学出版社, 2002: 94-127.
SU Ke-man, PAN Tie-ying, ZHANG Yu-lan, et al. Spectral analysis method[M]. Shanghai: East China University of Science and Technology Press, 2002: 94-127.
[13] 翁诗甫. 傅里叶变换红外光谱分析[M]. 北京: 化工工业出版社, 2010: 323-325.
WENG Shi-pu. Fourier transform infrared spectroscopy[M]. Beijing: Chemical Industry Press, 2010: 323-325.
[14] Yang Y. Adsorption of lysine on na-montmorillonite and competition with Ca2+: A combined XRD and ATR-FTIR study[J].Langmuir, 2016.
[15] 冯晓琴. 乌洛托品制备RDX硝解机理的理论研究[D]. 太原: 中北大学, 2008.
FENG Xiao-qin. The theoretical studies on nitrolysis mechanism of preparing RDX from urotropine[D]. Taiyuan: North University of China, 2008.
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
云南化工(2019年10期)2019-12-03 02:34:48
首都食品与医药(2019年22期)2019-10-24 10:20:36
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
腐蚀与防护(2018年12期)2018-12-20 02:31:06
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年1期)2014-02-28 17:30:06
