
典型难治性垂体腺瘤1例报告☆
2017-04-21 01:36刘小海代从新孙博文王任直
中国神经精神疾病杂志 2017年1期
刘小海代从新孙博文王任直
·病例报告·
典型难治性垂体腺瘤1例报告☆
刘小海*代从新*孙博文*王任直*
垂体腺瘤 难治性垂体腺瘤 非典型垂体腺瘤 垂体腺癌
大部分垂体腺瘤(pituitary adenoma,PA)表现为良性肿瘤的病程,但是临床上仍然会遇到一些处理棘手、最终无法挽救患者生命的病例。本文报道1例垂体腺瘤,术前肿瘤较小,规则生长,Knosp分级1-2级,符合非侵袭垂体腺瘤(non-invasive PA,NIPA),术后病理分型也未达到“非典型垂体腺瘤(atypical PA,APA)”诊断标准。但是,第一次手术全切后,肿瘤短时间内(小于半年)复发,且复发后生长速度明显加快。之后三次手术,联合放疗、常规化疗,仍然无法控制肿瘤生长,患者因高热、肺部感染及心肺功能衰竭死亡。希望以此来说明侵袭型垂体腺瘤及2004年WHO垂体腺瘤分型的不足,并探讨提示患者预后的“难治性垂体腺瘤(refractory PA,RPA)”的具体定义。
1 临床资料
1.1 一般资料患者,男,46岁,2010年4月体检时头颅CT提示鞍区占位,当时无明显不适,查垂体激素正常,考虑无功能垂体腺瘤,未进一步处理。2011年1月出现视力下降,自觉视野缩小,无明显内分泌系统症状体征,鞍区MRI显示垂体大腺瘤伴囊性变,大小约23mm×21mm×16mm,规则生长,Knosp分级1-2级(图1A)。
1.2 治疗过程及转归2011年1月26日于外院行内镜下经单鼻孔蝶窦入路垂体腺瘤切除术,术中肿瘤全切除,出现脑脊液漏,予自体肌肉修补。术后复查磁共振提示肿瘤全切除(影像资料丢失),术后视力视野明显改善,病理报告示:嫌色垂体腺瘤,免疫组化染色提示部分细胞PRL(+)、GH(+)、ACTH(+)、FSH(-)、LH(-)、Syn(+)及p53(-),同时Ki-67约1%。
2011年4月(术后3个月),患者再次出现双眼颞侧偏盲,复查鞍区MRI提示肿瘤复发,大小约22mm×20mm× 17mm,肿瘤包绕右侧海绵窦,侵袭左侧海绵窦(图1B)。查血促肾上腺皮质激素(adrenocorti cotrophic hormone,ACTH)为64pg/mL(正常<46 pg/mL),血总皮质醇正常范围,患者无库欣综合征相关症状和体征。考虑无功能垂体腺瘤术后复发,2011年5月16日于外院行右额开颅鞍区复发垂体腺瘤切除术,术中见肿瘤质软,位于视交叉下,视交叉被挤压向后移位,与左侧视神经粘连较重,肿瘤大部分切除(图1C)。术后免疫组化显示:ACTH(+),PRL(-)、GH(-)、FSH(-)、LH(-)、Syn(+)ChrA(+)及p53(-),Ki-67约10%。术后患者双眼视力视野改善,复查ACTH 15.30pg/mL,血总皮质醇5.17μg/dL。术后患者出现尿崩及甲状腺功能低下,口服优甲乐、强的松行激素替代治疗。
2011年10月12日(第二次开颅术后5个月)复查MRI,显示肿瘤体积明显增加,向两侧海绵窦及三脑室侵袭生长(图1D)。同时,患者开始出现满月脸、水牛背、血压升高等库欣综合征典型临床表现(图1L)。2012年4月13日,复查鞍区MRI显示肿瘤较前进一步增大,提示肿瘤细胞增殖加快(图1E)。
2012年8月3日,患者首次来我院就诊,鞍区MRI显示肿瘤较4个月前进一步增大,大小约30mm×25mm×23mm,包绕双侧海绵窦(图1F),查血皮质醇和ACTH分别达到1165 nmol/L和90pg/mL,小剂量地塞米松抑制试验不被抑制,大剂量可被抑制,考虑为垂体ACTH大腺瘤。2012年10月18日,患者于我院再次行内镜辅助下经鼻蝶窦入路垂体腺瘤切除术,术中发现鞍内一大小约2cm×2cm的腔隙,切除此处肿瘤,但脑干前方肿瘤位于鞍背后上方的桥前池内,考虑切除风险较大,未勉强切除(图1G),术后病理结果提示ACTH(+),PRL(-)、GH(-)、FSH(-)、LH(-)及p53(-),肿瘤细胞核分裂相明显增多,Ki-67约15%。
患者出院1月后,出现癫痫大发作,药物控制良好后,于我院放疗科进行分次立体定向放射治疗(病灶区28次,共50 Gy),症状无明显改善。期间应用溴隐亭(5mg,每日3次)2个月后检测血总皮质醇、ACTH轻度升高,提示溴隐亭治疗无效。2013年10月,患者再次出现头痛、视力视野减退伴右侧上睑下垂,MRI显示肿瘤生长迅速,大小30mm×25mm×27mm(图1H)。2014年3月21日,再次复查MRI,显示肿瘤进一步扩展(图1I)。2014年4月,患者头痛、视力障碍等症状进一步加重,于我院行开颅第四次手术,病理结果提示ACTH(+),PRL(-)、GH(-)、FSH(-)、LH(-)及p53(+),肿瘤细胞核分裂相多,Ki-67约20%(图2)。术后患者头痛和视力障碍部分改善(图1J),但1个月后上述症状迅速恶化,伴神志淡漠,寡语少动,小便失禁。查头CT发现患者出现明显的梗阻性脑积水(图1K),行脑室-腹腔分流术。术后头痛、神志淡漠等脑积水症状明显改善,复查头CT提示侧脑室三脑室均有所缩小,周边渗出改善。然而,肿瘤仍在迅速增大,2014年10月,患者因肿瘤占位、高皮质醇状态引起的高热、肺部感染及心肺功能衰竭去世。

图1 历次影像学检查。A:2011-1-19(第一次术前)MRI显示垂体大腺瘤伴囊性变,大小约23 mm×21 mm×16 mm;B:2011-4-27(第一次术后三个月)MRI显示肿瘤复发,大小约22 mm×20 mm×17 mm,肿瘤包绕右侧海绵窦,侵袭左侧海绵窦;C:2011-5-27(第二次术后):MRI显示术后肿瘤大部分切除;D:2011-10-12(第二次术后5个月)MRI显示肿瘤体积较术后明显增长,向两则海绵窦及三脑室侵袭生长;E:2012-4-13(第二次术后11个月)MRI显示肿瘤进一步增大;F:2012-8-3(第二次术后15个月)MRI显示肿瘤较4个月前进一步增大,大小约30 mm×25 mm×23 mm,包绕双侧海绵窦;G:2012-10-25(第三次术后)MRI显示鞍内肿瘤大部分切除,但脑干前方肿瘤位于鞍背后上方的桥前池内,考虑切除风险较大,未勉强切除;H:2013-10-31(第三次术后12个月)MRI肿瘤较第三次术后复查生长迅速,大小30 mm×25 mm×27 mm;I:2014-3-21(第三次术后17个月)MRI显示肿瘤进一步扩展;J:2014-4-24(第四次术后16d)MRI显示肿瘤大部分切除;K: 2014-5-5及5-28(第四次术后28d及50d)脑积水脑室腹腔分流术前后对比;L:病人库欣临床表现:肥胖、满月脸、痤疮、多血质面容等
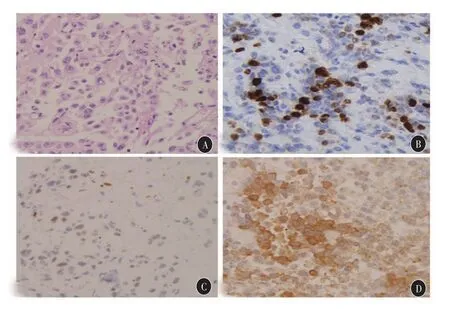
图2 第四次手术后病理检查 A:HE染色,400×;B:Ki-67约20%,400×;C:p53(+),400×;D:IHC示ACTH(++),400×
2 讨论
垂体腺瘤自然病程分型最常用的是2004年WHO分型,可分为:典型性垂体腺瘤(typical PA,TPA)、非典型性垂体腺瘤(APA)和垂体腺癌(pituitary carcinoma,PC)[1]。APA是指术后病理提示Ki-67抗原标记指数>3%、细胞核P53免疫组织化学染色阳性和/或细胞核异型性(细胞核分裂像增加)的垂体腺瘤,提示肿瘤增殖活性较高[2],病人预后不佳。如垂体腺瘤发生了蛛网膜下腔或者全身其他系统的转移,则称为垂体腺瘤(pituitary carcinoma,PC)[3]。PC非常罕见,文献报道只占所有垂体腺瘤的0.1%左右[4]。大多数PC是由反复手术治疗的垂体腺瘤或经过放疗的侵袭性腺瘤发展而来。
本例患者在第一次手术前考虑为无功能垂体腺瘤,手术全切,术后Ki-67约1%,符合典型性垂体腺瘤,提示预后良好。但在手术全切3个月后发现肿瘤复发,体积增大至原先大小,可见生长速度较前增快。患者又历经三次手术治疗和一次放射治疗,每次手术肿瘤大部分切除。但手术及放疗后,肿瘤生长加速,Ki-67指数逐步增高,且在最后一次手术病理中,p53转为(+),再次证实了垂体腺癌的发生来源之一[5]:初发为良性垂体腺瘤,但是经过多轮治疗后,肿瘤细胞进一步发生新的基因突变或进化,逐步转变为恶性的垂体腺癌,符合Knudson癌症的“二次打击学说”(two-hit hypothesis)。因此,在本例患者中,我们怀疑,肿瘤在多次手术后生长明显加速,多次病理提示细胞核分裂相及Ki-67指数递增,肿瘤有垂体腺癌的可能。在第四次术前,我们曾进行了全脊髓MRI等及其他系统检查,均未有发现蛛网膜下腔或其他系统的转移,因此,患者未能诊断为垂体腺癌。患者在第四次术后四个月因高热、肺部感染及心肺功能衰竭而死亡。
综上所述,该例病人虽符合“非侵袭垂体腺瘤”和“典型垂体腺瘤”标准,但多次手术,联合放疗、常规化疗,仍然无法控制肿瘤生长,其预后与垂体腺癌无异。因此,如何早期发现并准确定义这种类型的腺瘤,成为当务之急。总结既往病例,本课题组在国际上首次明确提出了难治性垂体腺瘤的定义:①肿瘤生长速度较快,Ki-67指数>3;②即使手术全切,肿瘤短期(半年内)复发;③手术、药物治疗和放疗均不能控制肿瘤生长,患者预后较差;④全身检查未见蛛网膜下腔或其他系统的转移[6]。
本例病例,肿瘤在第一次手术后,肿瘤类型发生了转变:由无功能垂体腺瘤转变为垂体ACTH腺瘤。复习文献,共有15篇类似报道[7]。此种转变的原因之一,可能是肿瘤本身为混合型或者单纯分泌型肿瘤,只是相关分泌激素未超过临床诊断标准并引发相关临床表现,术后病理未能检测到相关激素[7]。另一种原因考虑为针对原先肿瘤的放疗引发了肿瘤细胞内新的基因突变,相互叠加影响后,肿瘤类型发生迁跃[8]。本例肿瘤类型改变发生在放疗前,故可以排除第二种原因,第一种原因可能性大。此外,ACTH升高发生在临床症状体征之前,有很大意义。
针对难治性垂体腺瘤,目前尚无良好的诊断分子标志物。就目前病理检测而言,Ki-67增殖指数仍是最重要的内容之一,能初步判断肿瘤增殖活力,对预后判断有一定价值[9]。
[1]刘小海,冯铭,王任直.垂体腺瘤分型的历史、现状及展望[J].中国神经精神疾病杂志,2016,42(9):565-568.
[2]Al-SHRAIM M,ASA S.The 2004 world health organization clas⁃sification of pituitary tumors:what is new?[J].Acta Neuropathol, 2006,111(1):1-7.
[3]HEANEY AP.Clinical review:Pituitary carcinoma:difficult diag⁃nosis and treatment[J].J Clin Endocrinol Metab,2011,96(12): 3649-3660.
[4]HANSEN TM,BATRA S,LIM M,et al.Invasive adenoma and pi⁃tuitary carcinoma:a SEER database analysis[J].Neurosurg Rev, 2014,37(2):279-285.
[5]SUHARDJA AS,KOVACS KT,RUTKA JT.Molecular pathogene⁃sis of pituitary adenomas:a review[J].Acta Neurochirurgica, 1999,141(7):729-736.
[6]CONGXIN DAI,MING FENG,XIAOHAI LIU,et al.Refractory pituitary adenoma:a novel classification for pituitary tumors[J]. Oncotarget,2016,7(50):83657-83668,
[7]GHERVAN CMV,NEMES C,FLORIAN SI,et al.Silent cortico⁃troph adenoma transformed in secreting adenoma with severe Cushing’s disease after two pituitary surgeries[J].Acta Endo (Buc),2014,10(6):283-292.
[8]SANO T,KOVACS K,ASA SL,et al.Pituitary adenoma with‘honeycomb Golgi’appearance showing a phenotypic change at recurrence from clinically nonfunctioning to typical Cushing dis⁃ease[J].Endocr Pathol,2002,13(2):125–130.
[9]SALEHI F,AGUR A,SCHEITHAUER BW,et al.Ki-67 in pitu⁃itary neoplasms:a review--part I[J].Neurosurgery,2009,65(3): 429-437.
R730.26(
2016-08-17)
A(责任编辑:甘章平)
10.3969/j.issn.1002-0152.2017.01.012
☆ 十二五科技支撑项目(编号:BAIO4B03);国家自然科学基金青年项目(编号:81501192);2016年协和中青年科研基金(编号:pumch-2016-2.20)
*中国医学科学院,北京协和医学院,北京协和医院转化医学中心,北京协和医院神经外科,北京协和医院垂体诊治中心,卫生部内分泌重点实验室(北京 100730)
(E-mail:wangrz@126.com)
猜你喜欢
中国临床医学影像杂志(2022年2期)2022-05-25
世界科学技术-中医药现代化(2022年2期)2022-05-25
中国医学影像技术(2022年2期)2022-03-01
中老年保健(2021年5期)2021-08-24
中老年保健(2021年6期)2021-08-24
中老年保健(2021年11期)2021-08-22
天津医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2020年12期)2021-01-26
中华养生保健(2020年3期)2020-11-16
中国医学影像技术(2020年9期)2020-10-22
