
非甾体抗炎药洛索洛芬钠的合成
2017-04-14 10:27:06吴朝刚周雄飞庄程翰沈杭州张兴贤
合成化学 2017年4期
吴朝刚, 周雄飞, 庄程翰, 沈杭州, 张兴贤*
(1. 浙江普洛家园药业有限公司,浙江 东阳 322118; 2. 浙江工业大学 药学院, 浙江 杭州 310032)
·制药技术·
非甾体抗炎药洛索洛芬钠的合成
吴朝刚1, 周雄飞1, 庄程翰1, 沈杭州2, 张兴贤2*
(1. 浙江普洛家园药业有限公司,浙江 东阳 322118; 2. 浙江工业大学 药学院, 浙江 杭州 310032)
以1-苯基-2-氯-1-酮为原料,经缩酮保护、重排、水解、酯化及氯甲基化等反应制得关键中间体2-(4-氯甲基苯基)丙酸甲酯(7); 7与2-乙氧羰基环戊酮缩合后,再经脱羧及成盐反应合成了洛索洛芬钠,总收率36%,其结构经1H NMR和MS(ESI)确证。
1-苯基-2-氯-1-酮; 氯甲基化反应; 非甾体抗炎药; 洛索洛芬钠; 药物合成
洛索洛芬钠(1),化学名为2-[4-(2-氧代环戊基甲基)苯基]丙酸钠二水合物,由日本三共公司研发,于1986年首次在日本上市,临床用于类风湿性关节炎、腰痛、肩周炎、颈肩腕综合症等的抗炎镇痛、手术、外伤及拔牙后的镇痛消炎和急性上呼吸道炎症的解热镇痛等[1]。
1的合成方法较多[2-7],基本上需要经溴化反应制备2-(4-溴甲基)苯基丙酸甲酯来完成,存在反应操作繁琐、收率低、成本较高,需使用大量的金属盐催化剂和环境污染大等缺点,工业化生产具有一定局限性。
本文参考文献[3,8]方法,设计了一种1的8步合成法。以2-氯代苯丙酮(2)为起始原料,经新戊二醇保护、酸催化重排、碱性水解制得2-苯基丙酸(5); 5经甲酯化反应制得2-苯基丙酸甲酯(6); 6经氯甲基化反应制得关键中间体2-(4-氯甲基)苯基丙酸甲酯(7); 7在碱性条件下与2-乙氧羰基环戊酮缩合后,在酸性条件下水解脱羧得洛索洛芬酸(9); 9经成盐反应合成1,总收率36%,其结构经1H NMR和MS(ESI)确证。
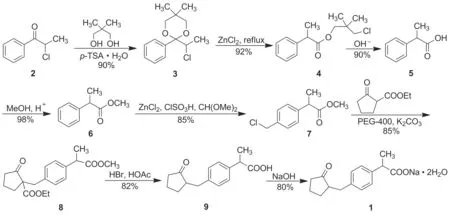
Scheme 1
1 实验部分
1.1 仪器与试剂
WRS-1A型数字熔点仪(温度未校正);Bruker AV 400型核磁共振仪(CDCl3为溶剂,TMS为内标);FTIR-8400S型红外光谱仪(液膜法);Finigan LCQ-MS型液质联用仪;Agilent 1200型高效液相色谱仪[色谱条件:色谱柱Kromasil C18柱(4.6 mm×250 mm, 5 μm),流动相:乙腈/水(V/V=60/40),检测波长:222 nm,柱温:25 ℃,流速:1.0 mL·min-1]; Agilent GC7890型气相色谱仪[色谱条件:色谱柱SPB-50 (0.25 mm×30.0 m, 0.25 m),柱温280 ℃,进样口温度260 ℃,程序升温(100 ℃保持2 min,以15 ℃·min-1升至220 ℃,保持15 min,再以30 ℃·min-1升至280 ℃,保持15 min)]。
所用试剂均为分析纯。
1.2 合成
(1) 2-(1-氯乙基)-5,5-二甲基-2-苯基-1,3-二氧六环(3)的合成
将2 10.0 g(59.36 mmol)溶于甲苯(120 mL)中,搅拌下依次加入新戊二醇10.5 g(100.88 mmol)和对甲苯磺酸2.28 g(12 mmol),回流(140 ℃)反应9 h(TLC跟踪)。自然冷却至室温,依次用水和饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩得黄色液体3 13.57 g,收率90%;1H NMRδ: 0.51(s, 3H, CH3), 1.27(s, 3H, CH3), 1.37(d,J=6.9 Hz, 3H, CH3), 3.40~3.43(m, 4H, CH2), 3.99(q,J=6.7 Hz, 1H, CHCl), 7.28~7.30(m, 1H, ArH), 7.33~7.36(m, 2H, ArH), 7.41~7.43(m, 2H, ArH)。
(2) 3-氯-2,2-二甲基丙基(2-苯基)丙酸酯(4)的合成
将3 10.0 g(39.3 mmol)溶于甲苯(50 mL)中,搅拌下加入ZnCl21.6 g(14.3 mmol),回流(130 ℃)反应6 h(TLC跟踪)。自然降至室温,依次用水和饱和食盐水洗涤,无水硫酸钠干燥,浓缩得淡黄色液体4 9.2 g,收率92%;1H NMRδ: 0.89(s, 3H, CH3), 0.93(s, 3H, CH3), 1.53(d,J=7.3 Hz, 3H, CH3), 3.26(dd,J=10.8 Hz, 12.8 Hz, 2H, CH2), 3.68(q,J=7.2 Hz, 1H, CHCH3), 3.87(d,J=11.0 Hz, 1H, CH), 3.99(d,J=11.0 Hz, 1H, CH), 7.24~7.28(m, 1H, ArH), 7.32~7.37(m, 4H, ArH)。
(3) 5的合成
将4 20.0 g(78.6 mmol)溶于甲醇(50 mL)中,搅拌下加入30%KOH水溶液(30 mL),回流(75 ℃)反应3 h(TLC跟踪)。自然冷却至室温,用浓盐酸调至pH 2~3,用EA(2×100 mL)萃取,合并萃取液,依次用水和饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩得淡黄色液体5 10.6 g,收率90%,纯度96%(GC,归一化法);1H NMRδ: 1.15(d,J=6.0 Hz, 3H, CH3), 3.68(q,J=6.0 Hz, 1H, CHCH3), 7.13~7.20(m, 5H, ArH); IRν: 3 322, 2 981, 2 976, 2 864, 1 705, 1 514, 1 456, 1 412 cm-1。
(4) 6的合成
将5 30.0 g(0.2 mol)和甲醇100 mL加入反应瓶中,搅拌使其均匀;于0~5 ℃滴加浓硫酸39.0 g(0.4 mol),滴毕,于室温反应6 h。减压蒸除约70 mL甲醇,加入水20 mL,用甲苯(2×60 mL)萃取,合并萃取液,依次用饱和碳酸钠溶液(20 mL)和水洗涤至中性,浓缩得淡黄色油状液体6 32.0 g,收率98%,纯度97%(GC,归一化法);1H NMRδ: 1.47(d,J=7.5 Hz, 3H, CH3), 3.72(q,J=7.5 Hz, 1H, CHCH3), 4.45(s, 3H, OCH3), 7.26~7.33(m, 5H, ArH); IRν: 3 028, 2 982, 2 951, 2 877, 1 736, 1 514, 1 454, 1 435, 1 421 cm-1。
(5) 7的合成
向250 mL三口瓶中加入无水氯化锌3.3 g(24.4 mmol)和氯磺酸35.0 g(0.304 mol),搅拌下缓慢滴加二甲氧基甲缩醛23.0 g(0.304 mol),滴毕,控制温度在-10 ℃以下,滴加6 20.0 g(0.122 mol),滴毕,自然升至室温,反应7 h。缓慢倒入冰水混合物(60.0 g)中,搅拌15 min,用甲苯(2×60 mL)萃取,合并萃取液,依次用10wt%碳酸钠水溶液(20 mL)、水(40 mL)和饱和氯化钠溶液(40 mL)洗涤,减压蒸除溶剂,油泵减压蒸馏,收集110~130 ℃/133 Pa馏分得无色油状液体7 22.0 g,收率85%,纯度80%(GC,归一化法),不需纯化直接用于下步反应。
(6) 2-[4-(1-乙氧羰基-2-氧代-1-环戊基甲基)苯基]丙酸甲酯(8)的合成
将2-乙氧羰基环戊酮23.4 g(0.15 mol)和甲苯100 mL加入反应瓶中,搅拌均匀,加入碳酸钾20.7 g(0.15 mol),升温至回流,滴加7 21.2 g(0.1 mol)的甲苯(50 mL)溶液,滴毕,回流反应12 h。冷却至室温,加入水80 mL,分液,水相用8.2 mol·L-1盐酸(15 mL)调至pH 3.0~4.0,用甲苯(100 mL)萃取,合并萃取液,用水洗涤,减压蒸除甲苯得黄色油状液体8 30.2 g,收率91%,纯度80%(HPLC,面积归一化法);1H NMRδ: 1.25(t,J=7.5 Hz, 3H, CH3CH2), 1.47(d,J=7.0 Hz, 3H, CH3), 1.63~1.65(m, 1H, CH2CH2), 1.89~1.96(m, 2H, CH2CH2, CH2CO), 2.03~2.07(m, 1H, CH2CO), 2.39~2.43(m, 2H, CH2CCO), 3.06(d,J=14.0 Hz, 1H, CH2Ar), 3.19(d,J=14.0 Hz, 1H, CH2Ar), 3.65(s, 3H, OCH3), 3.68(q,J=7.5 Hz, 1H, CHCH3), 4.16(q,J=7.5 Hz, 2H, OCH2), 7.09(d,J=8.0 Hz, 2H, ArH), 7.18(d,J=8.0 Hz, 2H, ArH); IRν: 2 976, 2 955, 2 889, 1 732, 1 514, 1 452, 1 435, 841 cm-1。
(7) 9的合成
将8 30 g(0.09 mol)、 48wt%氢溴酸60 mL和冰醋酸26 mL加入反应瓶中,回流反应8 h。冷却至室温,加入水50 mL,用甲苯(2×100 mL)萃取,萃取液用饱和氯化钠溶液洗涤,加入活性炭0.2 g脱色,过滤,在冰水浴中用乙酸乙酯/正己烷(V/V=1/2)搅拌析晶,过滤,干燥得白色固体9 15.5 g,收率70%,纯度99%(HPLC,面积归一化法), m.p.108~110 ℃(108.5~111 ℃[7,9]);1H NMRδ: 1.49(d,J=7.0 Hz, 3H, CH3), 1.51~1.56(m, 1H, CH2CH2), 1.70~1.74(m, 1H, CH2CH2), 1.94~1.98(m, 1H, CH2CO), 2.07~2.13(m, 2H, CH2CO, CH2CHCO), 2.31~2.34(m, 2H, CH2CHCO, CHCO), 2.51(dd,J=4.0 Hz, 14.0 Hz, 1H, CH2Ar), 3.13(dd,J=4.0 Hz, 14.0 Hz, 1H, CH2Ar), 3.70(q,J=7.0 Hz, 1H, CHCH3), 7.13(d,J=8.0 Hz, 2H, ArH), 7.23(d,J=8.0 Hz, 2H, ArH); IRν: 3 020, 2 966, 2 935, 2 889, 2 874, 1 734, 1 701, 1 512, 1 452, 1 421, 1 400 cm-1。
(8) 1的合成
将9 12.3 g(50 mmol)和乙醇50 mL加入反应瓶中,搅拌下于室温滴加20wt%氢氧化钠水溶液(10 mL)调至pH 7.0~8.0,滴毕,于室温反应3 h。析晶,过滤,用乙醇/异丙醚(V/V=1/1)重结晶得白色固体1 13.9 g(45.7 mmol),收率91.5%,纯度99%(HPLC,面积归一化法), m.p.196~198 ℃(197~199 ℃[3]);1H NMR(D2O)δ: 1.26(d,J=7.2 Hz, 3H, CH3), 1.44~1.53(m, 1H, CH2CH2), 1.59~1.69(m, 1H, CH2CH2), 1.80~1.91(m, 2H, CH2CO), 2.03~2.09(m, 1H, CH2CHCO), 2.21~2.26(m, 1H, CH2CHCO), 2.40~2.51(m, 2H, CHCO, CH2Ar), 2.88(dd,J=4.4 Hz, 13.5 Hz, 1H, CH2Ar), 3.60(q,J=7.5 Hz, 1H, CHCH3), 7.08(d,J=8.0 Hz, 2H, ArH), 7.15(d,J=8.0 Hz, 2H, ArH); MS(ESI)m/z: 245{[M-Na]-}。
在以往1的制备研究中,基本上采用2-(4-溴甲基)苯基丙酸作为关键中间体[3-5],该溴化物的合成使用溴素或N-溴代丁二酰亚胺(NBS)作为溴化试剂,通过光照或加热引发的方法对2-(4-甲基苯基)丙酸中的甲基进行溴化,由于溴化反应副产物较多,需多次精制和纯化,收率较低,生产成本较高,且后处理比较繁琐,操作上存在一定的安全隐患。
另外,文献[3-4]方法通过重排反应合成2-(4-甲基苯基)丙酸,需要使用化学计量的金属盐或单质碘作催化剂,环境污染较严重,工业化生产上存在一定局限性。文献[10]方法通过贵金属钯催化的烯烃羰基化反应来制备2-(4-甲基苯基)丙酸,需要使用昂贵的贵金属催化剂和4-甲基苯乙烯为原料,反应条件苛刻,工业化生产成本高。
本文设计了一条新的合成工艺路线,从价廉、易得的2出发,经缩酮保护、重排、水解、酯化、氯甲基化等反应制得关键中间体7; 7与2-乙氧羰基环戊酮经缩合、脱羧、成盐得目标产物1,总收率36%。其中,关键中间体5的制备通过无水氯化锌催化的Blanc氯甲基化反应实现,避免溴素的使用,产物不需要进一步纯化,可直接进入下一步缩合反应,从而简化了操作步骤,提高了生产效率;得到的洛索洛芬酸粗品经乙酸乙酯-正己烷重结晶,其纯度可以提高至99%,进而制得1。
综上所述,该合成路线较文献方法具有步骤简短,操作简便,生产成本低,反应条件温和等优点,为洛索洛芬钠的研究和生产提供了一定的参考,具有较好的工业化应用前景。
[1] Kawano S, Tsuji S, Hayashi N,etal. Effects of loxoprofen sodium,a newly synthesized non-steroidal anti-inflammatory drug,and indomethacin on gastric mucosal haemodynamics in the human[J].Journal of Gastroenterology and Hepatology,1995,10:81-85.
[2] Maurizio S, Carios A M, Pietro T. Selective mono-methylation of aryacetonitriles and methyl arylacetates by dimethyl carbonate[J].Journal of Chemical Society:Perkin Trans I,1994,10:1323-1328.
[3] 陈芬儿,刘安昌,户业丽,等. 洛索洛芬钠的重排合成工艺研究[J].中国医药工业杂志,1998,29:531-533.
[4] 李爱军,周雪琴. 碘催化芳基重排法合成2-(4-溴甲基苯基)丙酸[J].精细化工,2006,23:613-614.
[5] 刘小帆,邓建成,申东升. 2-(4-溴甲基苯基)丙酸乙酯的合成[J].合成化学,2005,13:526-528.
[6] Curini M, Epifano F, Maltese F,etal. Carbamate synthesis from amines and dimethyl carbonate under ytterbium triflate catalyst[J].Tetrahedron Letters,2002,43:4895-4897.
[7] 冯姣,潘鹤林,禹艳坤,等. 洛索洛芬钠的合成新工艺研究[J].化学试剂,2016,38:88-90.
[8] Terade A, Naruto S, Wachi K,etal. Synthesis and antiinflammatory activity of [(cycloalkylmethyl)phenyl]cetic acids and related compounds[J].Journal of Medical Chemistry,1984,27:212-216.
[9] Terade A, Wachi K, Misaka E,etal. Substituted phenylacetic acid derivatives and process for the preparation thereof:US 4161538[P].1979.
[10] Seayad A, Jayasree S, Chaudhari R V. Carbonylation of vinyl aromatics:Convenient regioselective synthesis of 2-arylpropanoic acids[J].Organic Letters,1999,1:459-462.
Synthesis of Nonsteroidal Anti-inflammatory Drug Loxoprofen Sodium
WU Chao-gang1, ZHOU Xiong-fei1, ZHUANG Cheng-han1, SHEN Hang-zhou2, ZHANG Xing-xian2*
(1. Zhejiang Apeloajiayuan Pharmaceutical Co., Ltd., Dongyang 322118, China; 2. College of Pharmacy, Zhejiang University of Technology, Hangzhou 310032, China)
The key intermediate, methyl 2-(4-chloromethylpheny1)propionate(7), was prepared by five steps of ketalization, rearrangement, hydrolysis, esterification and chloromethylation, using 1-phenyl-2-chloro-1-one as starting material. Loxoprofen sodium with overall yield of 36% was synthesized from 7 by phase-transfer catalyzed alkylation with ethyl 2-oxocyclopentanecarboxylate, and then decarboxylation and salt formation. The structure was confirmed by1H NMR and MS(ESI).
1-phenyl-2-chloro-1-one; chloromethylation; nonsteroidal anti-inflammatory drug; loxoprofen sodium; drug synthesis
2016-09-26;
2017-02-20
吴朝刚(1976-),男,苗族,贵州湄潭人,学士,主要从事药物合成的研究。 Tel. 0579-86558258, E-mail: chaogangwu@apeloa.com
张兴贤,教授, Tel. 0571-88320913, E-mail: zhangxx@zjut.edu.cn
O621.3; R914.5
A
10.15952/j.cnki.cjsc.1005-1511.2017.04.16246
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:52
农药科学与管理(2019年8期)2019-11-23 08:04:44
现代食品(2016年24期)2016-04-28 08:12:06
化工进展(2015年3期)2015-11-11 09:07:41
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国当代医药(2015年10期)2015-03-01 02:02:39
海军医学杂志(2015年2期)2015-02-27 13:47:35
化学工业与工程(2015年1期)2015-02-10 03:01:33
应用化工(2014年3期)2014-08-16 13:23:50
无机化学学报(2014年12期)2014-02-28 17:34:01
