
偕双硅2,3-环氧醇区域及立体选择性环氧开环/卤代反应研究
2017-04-14 10:27:03宋振雷
合成化学 2017年4期
胡 佳, 高 璐, 宋振雷
(四川大学 华西药学院,四川 成都 610041)
·研究论文·
偕双硅2,3-环氧醇区域及立体选择性环氧开环/卤代反应研究
胡 佳, 高 璐*, 宋振雷*
(四川大学 华西药学院,四川 成都 610041)
C3-位偕双硅取代的2,3-环氧醇分别与对甲苯磺酰氯、溴素和单质碘发生C3-位区域及立体选择性环氧开环/卤代反应合成了15个新的C3-位卤代偕双硅基1,2-二醇类产物,收率64%~87%,其结构经1H NMR,13C NMR和HR-MS(ESI)表征。产物的立体结构经3-[二甲基(苯基)甲硅烷基]-3-碘-3-(三甲基甲硅烷基)丁烷-1,2-二醇(8d)的X-射线单晶衍射确证。
偕双硅; 2,3-环氧醇; 环氧开环; 卤代反应; 区域选择性; 立体选择性; 合成
2,3-环氧醇是一类具有广泛应用价值的有机合成砌块,可通过烯丙醇的环氧化反应方便制得。该类化合物的一个重要转化是发生环氧开环反应,生成双羟基化合物。由于环氧开环既可以发生在C2-位生成1,3-双羟基化合物,也可以发生在C3-位生成1,2-双羟基化合物,因此控制反应的区域选择性显得非常关键。这也是2,3-环氧醇化学长期以来一个重要且具有挑战性的课题。目前,实现C3-位选择性开环的代表性工作主要有:(1)金属离子螯合作用介导的亲核试剂环氧开环反应[1-2];(2)路易斯酸催化的亲核试剂环氧开环反应[3-5];(3)钨、锰和铯等金属盐促进的环氧开环反应[6-7];四、有机硼催化的氯代酰化反应[8]。但是,当C3-位为双取代且取代基体积较大时,实现C3-位的高区域选择性环氧开环则十分困难,有关这方面的研究工作至今鲜有报道。
为了探索C3-位有大体积硅基团取代的2,3-环氧醇的开环区域选择性,同时基于本课题组对偕双硅化学的深入研究,本文以3-丁炔-2-醇为起始原料,通过几步转化合成了5种C3-位偕双硅取代的2,3-环氧醇化合物(5a~5e, Scheme 1,以5a的合成为例)[9-13]; 5a~5e分别与TsCl、Br2和I2发生C3-位区域及立体选择性环氧开环/卤代反应,合成了15个新的C3-位卤代偕双硅基1,2-二醇类产物(6a~6e, 7a~7e和8a~8e),收率64%~87%(Scheme 2),其结构经1H NMR,13C NMR和HR-MS(ESI)表征。产物的立体化学经3-[二甲基(苯基)甲硅烷基]-3-碘-3-(三甲基甲硅烷基)丁烷-1,2-二醇(8d)的X-射线单晶衍射确证。首次证实了当2,3-环氧醇的C3-位为大体积的硅基团取代时,在碱性条件下的环氧开环/氯代反应区域选择性地发生在大位阻的C3-位,而非位阻更小的C2-位。
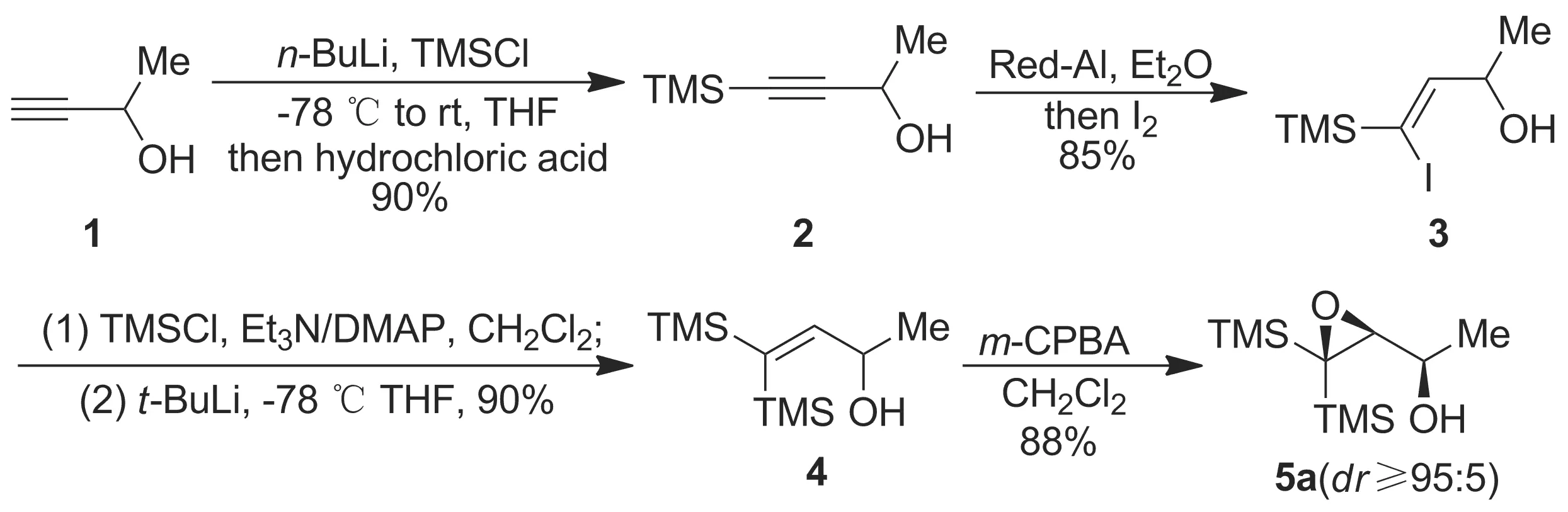
Scheme 1
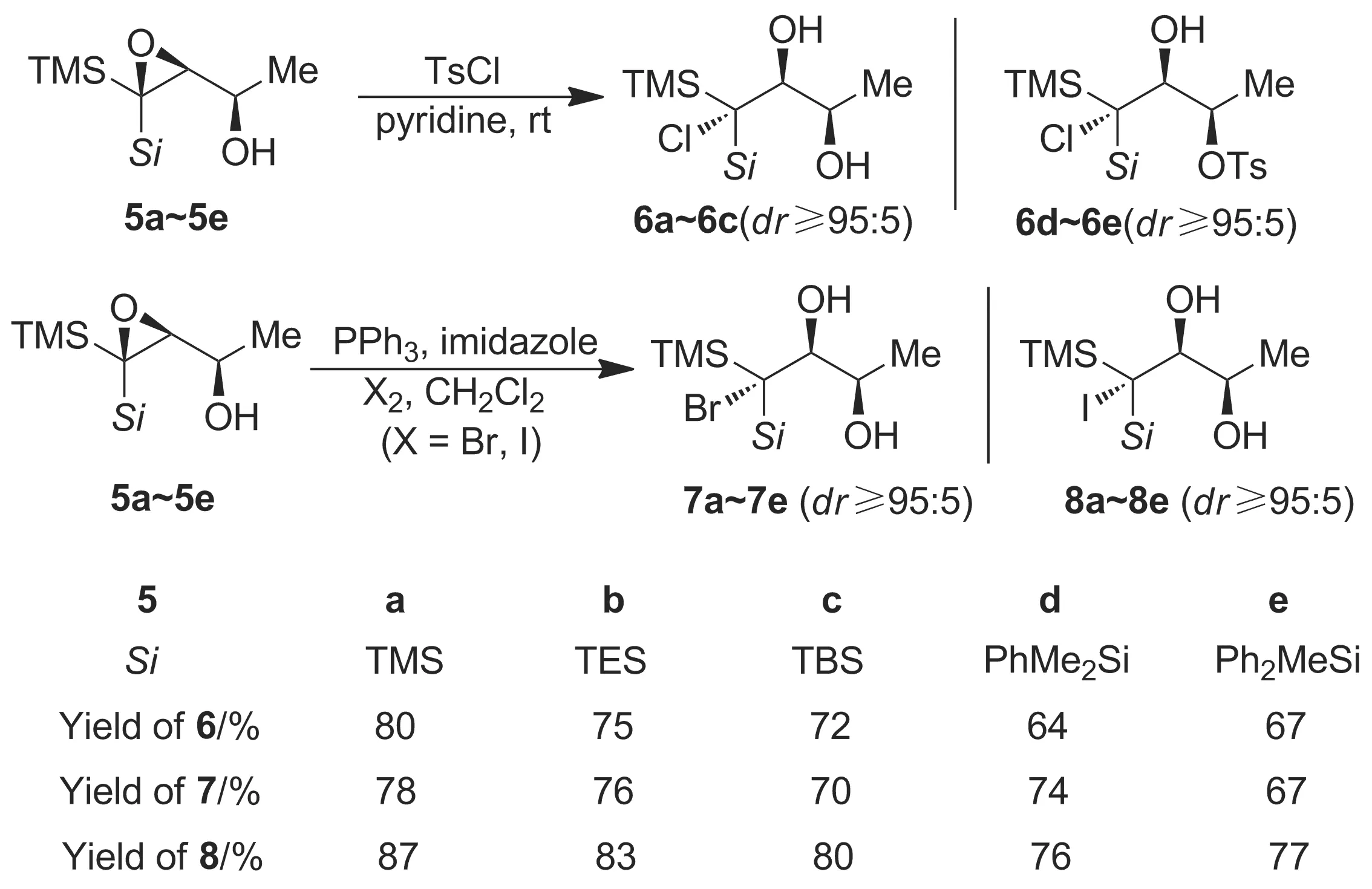
Scheme 2
1 实验部分
1.1 仪器与试剂
Varian Unity NOVA-400 MHZ/54型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters Q-TOF-Premier型质谱仪。
5a~5e参照文献[9-13]方法合成;硅胶,200~300目,青岛海洋化工厂;GF254硅胶板,烟台江友硅胶开发有限公司;其余所用试剂均为分析纯。
1.2 合成
(1) 6a~6e的合成(以6a为例)
氩气保护下,在反应管中加入5a 50 mg(0.21 mmol)和新蒸吡啶1 mL,搅拌使其混合均匀;随后向体系中加入对甲苯磺酰氯45 mg(0.24 mmol),于室温反应24 h(TLC检测)。加入乙酸乙酯2 mL,依次用饱和NaHCO3溶液和饱和NaCl溶液洗涤,无水Na2SO4干燥,浓缩后粗产物经硅胶柱层析[洗脱剂:A=石油醚/乙酸乙酯=30/1,V/V]纯化得6a。
以5b~5e替代5a,用类似方法合成6b~6e。
6a: 无色油状液体,收率80%;1H NMRδ: 4.22~4.18(m, 1H), 3.78(d,J=9.0 Hz, 1H), 3.10(d,J=9.0 Hz, 1H), 2.15(d,J=3.6 Hz, 1H), 1.27(d,J=5.4 Hz, 3H), 0.23(s, 9H), 0.19(s, 9H);13C NMRδ:77.9, 67.2, 57.7, 21.5, 0.7, -0.3; HR-MS(ESI)m/z: Calcd for C10H25O2Si2Cl{[M+Na]+}291.097 4, found 291.096 1。
6b: 无色油状液体,收率75%;1H NMRδ: 4.19~4.15(m, 1H), 3.77(d,J=9.0 Hz, 1H), 3.25(d,J=8.4 Hz, 1H), 2.30(d,J=4.2 Hz, 1H), 1.25(d,J=6.0 Hz, 3H), 1.04(t,J=7.8 Hz, 9H), 0.92~0.77(m, 6H), 0.19(s, 9H);13C NMRδ: 77.4, 68.9, 57.3, 21.5, 8.9, 7.1, 0.7; HR-MS(ESI)m/z: Calcd for C13H31O2Si2Cl{[M+Na]+}333.144 3, found 333.147 1。
6c: 无色油状液体,收率72%;1H NMRδ: 4.23~4.19(dq,J=6.6 Hz, 1.8 Hz, 1H), 3.88(d,J=9.0 Hz, 1H), 3.36(d,J=9.0 Hz, 1H), 2.44(d,J=4.2 Hz, 1H), 1.26(d,J=6.6 Hz, 3H), 1.03(s, 9H), 0.24(s, 3H), 0.23(s, 9H), 0.18(s, 3H);13C NMRδ: 76.1, 68.7, 65.2, 28.9, 20.9, 20.3, 0.7, -1.4, -2.0; HR-MS(ESI)m/z: Calcd for C13H31O2Si2Cl{[M+Na]+}333.144 3, found 333.145 2。
6d: 无色油状液体,收率64%;1H NMRδ: 7.71(d,J=8.0 Hz, 2H), 7.45(d,J=6.4 Hz, 3H), 7.39~7.30(m, 5H), 4.26~4.19(m, 1H), 3.05(d,J=8.0 Hz, 1H), 2.44(s, 3H), 1.17(d,J=6.4 Hz, 3H), 0.35(s, 3H), 0.33(s, 3H), -0.06(s, 9H);13C NMRδ: 144.3, 137.3, 134.8, 134.2, 134.1, 129.6, 128.0, 127.9, 80.6, 63.7, 51.7, 21.6, 18.3, 0.3, -1.0, -2.2; HR-MS(ESI)m/z: Calcd for C22H33O4Si2SCl{[M+Na]+}507.121 9, found 507.122 0。
6e: 无色油状液体,收率67%;1H NMRδ: 7.80(d,J=8.0 Hz, 2H), 7.48(t,J=6.8 Hz, 4H), 7.41~7.29(m, 9H), 4.48(dq,J=6.4 Hz, 1.6 Hz, 1H), 2.89(d,J=7.2 Hz, 1H), 2.45(s, 3H), 1.45(d,J=6.4 Hz, 3H), 0.64(s, 3H), -0.12(s, 9H);13C NMRδ: 144.4, 135.0, 134.8, 134.4, 134.3, 134.1, 129.8, 129.6, 129.5, 128.0, 127.9, 127.8, 80.6, 63.4, 50.7, 21.6, 18.6, 1.0, 0.1, -4.3; HR-MS(ESI)m/z: Calcd for C27H35O4Si2SCl{[M+Na]+}546.148 3, found 546.148 7。
(2) 7a~7e的合成(以7a为例)
氩气保护下,在反应管中加入5a 50 mg(0.21 mmol)和新蒸二氯甲烷1 mL,搅拌使其混合均匀;依次加入三苯基膦62 mg(0.24 mmol),咪唑33 mg(0.48 mmol)和溴素0.17 mL(0.31 mmol, 0.1 mL Br2/1 mL CH2Cl2),于室温反应8 h(TLC检测)。加入乙酸乙酯2 mL,依次用饱和NaHCO3溶液和饱和NaCl溶液洗涤,无水Na2SO4干燥,浓缩,粗产物经硅胶柱层析(洗脱剂:A=30/1)纯化得7a。
以5b~5e替代5a,用类似方法合成7b~7e。
7a: 无色油状液体,收率78%;1H NMRδ: 4.24~4.20(m, 1H), 3.81(d,J=9.0 Hz, 1H), 3.12(d,J=9.0 Hz, 1H), 2.20(d,J=4.2 Hz, 1H), 1.26(d,J=6.6 Hz, 3H), 0.25(s, 9H), 0.23(s, 9H);13C NMRδ: 77.8, 67.6, 56.4, 21.7, 1.4, 0.5; HR-MS(ESI)m/z: Calcd for C10H25O2Si2Br{[M+Na]+}312.057 6, found 312.057 0。
7b: 无色油状液体,收率76%;1H NMRδ: 4.20~4.16(m, 1H), 3.77(d,J=8.8 Hz, 1H), 3.27(d,J=8.8 Hz, 1H), 2.37(d,J=3.6 Hz, 1H), 1.25(d,J=5.6 Hz, 3H), 1.05(t,J=8.0 Hz, 9H), 0.98~0.77(m, 6H), 0.22(s, 9H);13C NMRδ: 77.3, 68.5, 57.1, 21.4, 8.7, 6.2, 0.6; HR-MS(ESI)m/z: Calcd for C13H31O2Si2Br{[M+Na]+}377.093 8, found 377.094 0。
7c: 无色油状液体,收率70%;1H NMRδ: 4.20~4.17(m, 1H), 3.84(d,J=8.8 Hz, 1H), 3.35(d,J=8.8 Hz, 1H), 2.53(d,J=4.0 Hz, 1H), 1.25(d,J=6.0 Hz, 3H), 1.04(s , 9H), 0.27(s, 3H), 0.26(s, 9H), 0.20(s, 3H);13C NMRδ: 75.8, 69.4, 62.5, 29.0, 21.1, 20.7, 1.5, 0.4, -1.5; HR-MS(ESI)m/z: Calcd for C13H31O2Si2Br{[M+Na]+}377.093 8, found 377.094 0。
7d: 无色油状液体,收率74%;1H NMRδ: 7.86~7.83(m, 4H), 7.43~7.33(m, 6H), 4.06(d,J=9.2 Hz, 1H), 3.79~3.73(m, 1H), 3.27(d,J=9.2 Hz, 1H), 2.15(d,J=4.4 Hz, 1H), 0.94(s, 3H), 0.93(d,J=6.4 Hz, 3H), 0.03(s, 9H);13C NMRδ: 138.5, 135.4, 129.3, 127.5, 77.4, 67.9, 21.6, 0.5, 0.1, -0.1; HR-MS(ESI)m/z: Calcd for C15H27O2Si2Br{[M+Na]+}397.062 5, found 397.062 1。
7e: 无色油状液体,收率67%;1H NMRδ: 7.86~7.85(m, 4H), 7.44~7.33(m, 6H), 4.05(d,J=8.8 Hz, 1H), 3.77~3.73(m, 1H), 3.27(d,J=9.2 Hz, 1H), 2.15(d,J=4.0 Hz, 1H), 0.93(s, 3H), 0.92(d,J=8.8 Hz, 3H), 0.03(s, 9H);13C NMRδ: 136.1, 135.5, 135.1, 131.3, 129.9, 129.6, 128.0, 127.8, 76.7, 67.9, 21.1, 1.5, -0.7; HR-MS(ESI)m/z: Calcd for C20H29O2Si2Br{[M+Na]+}459.078 2, found 459.078 1。
(3) 8a~8e的合成(以8a为例)
氩气保护下,在反应管中加入5a 50 mg(0.21 mmol)和新蒸二氯甲烷1 mL,搅拌使其混合均匀;随后依次向体系中加入三苯基膦62 mg(0.24 mmol),咪唑33 mg (0.48 mmol)及碘粒82 mg(0.32 mmol),于室温反应6 h(TLC检测)。加入乙酸乙酯2 mL,依次用饱和NaHCO3溶液和饱和NaCl溶液洗涤,无水Na2SO4干燥,浓缩后粗产物经硅胶柱层析(洗脱剂:A=30/1)纯化得8a。
以5b~5e替代5a,用类似方法合成8b~8e。
8a: 淡黄色油状液体,收率87%;1H NMRδ: 4.29~4.23(m, 1H), 3.72(d,J=9.2 Hz, 1H), 3.14(d,J=9.2 Hz, 1H), 2.31(d,J=4.8 Hz, 1H), 1.24(d,J=6.4 Hz, 3H), 0.28(s, 9H), 0.26(s, 9H);13C NMRδ: 78.7, 68.1, 40.1, 22.3, 2.7, 1.8; HR-MS(ESI)m/z: Calcd for C10H25O2Si2I{[M+Na]+}383.033 0, found 383.033 1。
8b: 淡黄色油状液体,收率83%;1H NMRδ: 4.17~4.14(m, 1H), 3.58(d,J=8.8 Hz, 1H), 3.23(d,J=8.8 Hz, 1H), 2.33(s, 1H), 1.24(d,J=6.4 Hz, 3H), 1.06(t,J=7.2 Hz, 9H), 0.99~0.82(m, 6H), 0.26(s, 9H);13C NMRδ: 77.4, 69.5, 41.4, 21.9, 9.0, 7.4, 1.8; HR-MS(ESI)m/z: Calcd for C13H31O2Si2I{[M+Na]+}425.079 9, found 425.078 9。
8c: 淡黄色油状液体,收率80%;1H NMRδ: 4.14~4.08(m, 1H), 3.47(d,J=8.8 Hz, 1H), 3.27(d,J=8.8 Hz, 1H), 2.49(d,J=4.4 Hz, 1H), 1.23(d,J=6.4 Hz, 3H), 1.06(s, 9H), 0.32(s, 3H), 0.31(s, 9H), 0.24(s, 9H);13C NMRδ: 75.5, 70.2, 44.8, 29.2, 21.7, 21.2, 3.0, 1.6, -0.8; HR-MS(ESI)m/z: Calcd for C13H31O2Si2I{[M+Na]+}425.079 9, found 425.079 0。
8d: 白色固体,收率76%, m.p. 84~88 ℃;1H NMRδ: 7.78~7.77(m, 2H), 7.39~7.34(m, 3H), 4.19~4.17(m, 1H), 3.69(d,J=8.4 Hz, 1H), 3.24(d,J=9.0 Hz, 1H), 2.23(d,J=4.2 Hz, 1H), 1.23(d,J=6.0 Hz, 3H), 0.67(s, 3H), 0.65(s, 3H), 0.05(s, 9H);13C NMRδ: 138.8, 135.5, 129.3, 127.4, 77.6, 68.9, 41.1, 22.1, 2.6, 1.4, 0.7; HR-MS(ESI)m/z: Calcd for C15H27O2Si2I{[M+Na]+}445.048 6, found 445.048 1。
8e: 黄色油状液体,收率77%;1H NMRδ: 7.94(d,J=6.8 Hz, 2H), 7.90(d,J=6.8 Hz, 2H), 7.45~7.33(m, 6H), 3.89(d,J=8.8 Hz, 1H), 3.85~3.82(m, 1H), 3.27(d,J=8.8 Hz, 1H), 2.14(d,J=4.4 Hz, 1H), 1.02(s, 3H), 0.96(d,J=6.4 Hz, 3H), 0.06(s, 9H);13C NMRδ: 136.5, 136.1, 135.7, 134.3, 129.8, 129.5, 127.9, 127.7, 76.9, 68.7, 41.3, 21.7, 2.8, 2.4; HR-MS(ESI)m/z: Calcd for C20H29O2Si2I{[M+Na]+}507.064 3, found 507.064 1。
2 结果与讨论
2.1 反应适用范围
2,3-环氧醇5a~5e含有不同的偕双硅组合,其中反式的硅取代基均为TMS。当TsCl为氯源时,不同的顺式硅取代基的立体和电子效应对环氧开环的有效性并无明显影响。反应表现出了单一的C3-位环氧开环区域选择性,并以较好的收率获得6a~6e(Scheme 2),而且反应中未检测到C2-位环氧开环产物的生成。从产物6b~6e的立体化学可以判断出,氯离子的进攻方向应为环氧的背面。这种立体选择性和碱性条件下环氧SN2的开环方式是一致的。另外,这也在一定程度上排除了C3以碳正离子方式参与的可能性。实际上,由于硅的原子半径大于碳,所以相应的偕双硅取代的三级碳正离子通常并不具有碳取代三级碳正离子的稳定性。另外我们发现,对于5d和5e,反应生成的1,2-二醇可以继续发生区域选择性的单磺酰化,从而转化为6d和6e。这样,我们就在一步转化中同时实现了环氧开环和羟基选择性官能团化两种不同的区域选择性控制。
当Br2和I2为卤源时,2,3-环氧醇5a~5e还可以转化为相应的溴代二醇7a~7e,以及碘代二醇8a~8e(Scheme 2)。反应仍然表现出了单一的C3-位环氧开环的区域选择性,以及反式进攻的立体选择性。其中,通过8d的X-射线单晶衍射对产物的立体化学进行了确证(图1)。
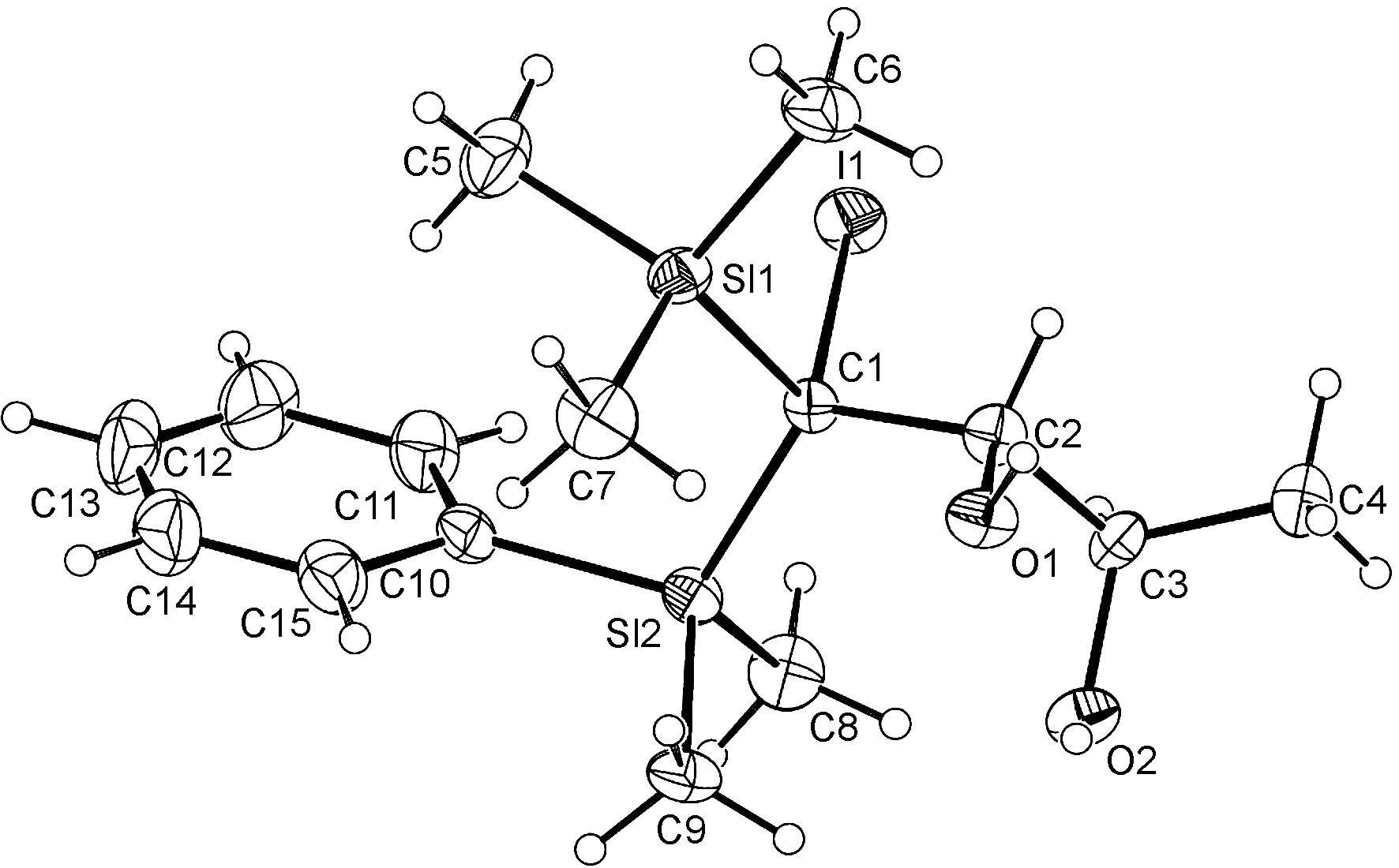
图1 8d的X-射线单晶衍射结构
2.2 反应机理
由于受两个体积庞大的硅的空间屏蔽效应影响,偕双硅取代的C3-位的空间位阻远大于C2-位。因此,在碱性条件下的环氧开环似乎更应该在C2-位而非C3-位发生。然而实际的反应结果却与预期恰恰相反,即卤代开环反应发生在C3-位。对于这种独特的区域选择性,我们给出了如下可能的推测。首先,卤素负离子与具有微弱路易斯酸性的硅中心有可能形成了类似五配位硅的中间体9(Chart 1)。这种邻基参与效应从而促使环氧开环选择性地发生在C3-位[14-15]。另外一个可能的原因是,硅基团的给电子效应和空间排斥造成的挤压效应有可能弱化了C3—O键,使之相较于C2—O键更易于断裂。
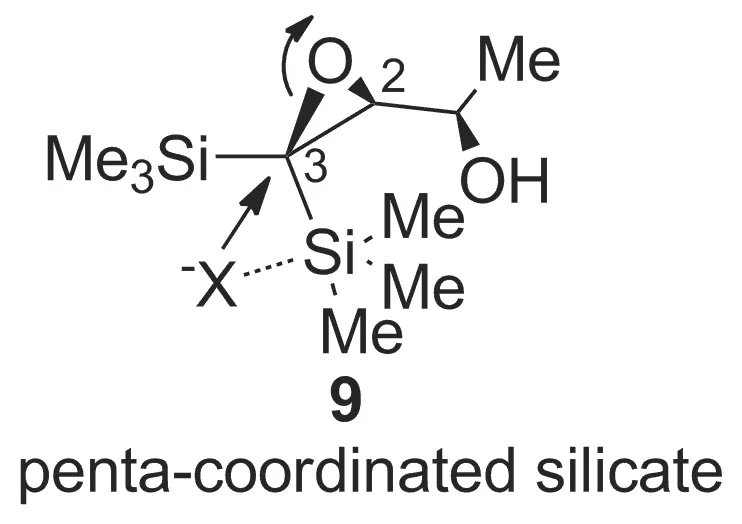
Chart 1
本工作详细研究了C3-位偕双硅取代的2,3-环氧醇在碱性条件下开环/卤代反的应区域选择性,以较好的收率(64%~87%)合成了15个C3位区域选择性环氧开环/卤代的产物。首次证实了当2,3-环氧醇的C3-位为大体积的硅基团取代时,在碱性条件下的环氧开环发生在大位阻的C3-位,而非位阻更小的C2-位。该研究工作既为2,3-环氧醇化合物的区域选择性开环反应提供了新的实例,同时也丰富了偕双硅在反应选择性控制方面的独特功能。
[1] Caron M, Sharpless K B. Ti(O-i-Pr)4-mediated nucleophilic openings of 2,3-epoxy alcohols.A mild procedure for regioselective ring-opening[J].J Org Chem,1985,50(9):1557-1560.
[2] Chini M, Crotti P, Flippin L A,etal. Regiochemical control of the ring opening of 1,2-epoxides by means of chelating processes.5.Synthesis and reactions of some 2,3-epoxy-1-alkanol derivatives[J].J Org Chem,1993,58(5):1221-1227.
[3] Maruoka K, Sano H, Yamamoto H. A highly region- and stereoselective ring-opening of 2,3-epoxy alcohols with trimethylsilyl azide-diethylaluminum fluoride system[J].Chem Lett,1985,14(5):599-602.
[4] Guivisdalsky P, Bittman R. Glycidyl derivatives as chiral C3 synthons,ring opening catalyzed by BF3etherate[J].J Am Chem Soc,1989,111(8):3077-3079.
[5] Uesugi S, Watanabe T, Imaizumi T,etal. Eu(OTf)3-catalyzed highly regioselective nucleophilic ring opening of 2,3-epoxy alcohols:An efficient entry to 3-substituted 1,2-diol derivatives[J].Org Lett,2014,16(17):4408-4411.
[6] Wang C, Yamamoto H. Tungsten-catalyzed regioselective and stereospecific ring opening of 2,3-epoxy alcohols and 2,3-epoxy sulfonamides[J].J Am Chem Soc,2014,136(19):6888-6891.
[7] Wang C, Yamamoto H. Tungsten-,molybdenum-,and cerium-promoted regioselective and stereospecific halogenation of 2,3-epoxy alcohols and 2,3-epoxy sulfonamides[J].Org Lett,2014,16(22):5937-5939.
[8] Kashif T, Kareem J, Mark S T. Borinic acid catalyzed,regioselective chloroacylations and chlorosulfonylations of 2,3-epoxy alcohols[J].Org Lett,2015,17(14):3482-3485.
[9] Corey E J, Katzenellenbogen J A, Posner G H. New stereospecific synthesis of trisubstituted olefins.Stereospecific synthesis of farnesol[J].J Am Chem Soc,1967,89(16):4245-4247.
[10] Denmark S E, Jones T K. (E)-3-(Trimethylsilyl)-2-propen-1-ol.An improved preparation[J].J Org Chem, 1982,47(23):4595-4597.
[11] Kim D K, Magtiotis P A. A new stereoselective synthesis of (Z)-vinylsilane allylic alcohols[J].Tetrahedron Lett,1990,31(43):6137-6140.
[12] Yan L J, Sun X W, Li H Z,etal. Geminal bis(silyl) enal:A versatile scaffold for stereoselective synthesizingC3,O1-disilylated allylic alcohols based upon anion relay chemistry[J].Org Lett,2013,15(5):1104-1107.
[13] Hodgson D M, Comina P J, Drew M G B. Chromium(II)-mediated synthesis synthesis of vinylbis(silanes) from aldehydes and a study of acid- and base-induced reactions of the derived epoxybis(silanes):A synthesis of acylsilanes[J].J Chem Soc,Perkin Trans 1,1997:2279-2289.
[14] Eisch J J, Chiu C S. Pentacoordinate silicon intermediates in relay substitution reactions of organosilanes:Successive nucleophilic attack at silicon and its adjacent carbon[J].Journal of Organometallic Chemistry,1988,358:C1-C5.
[15] Michio O, Kiitiro U, Hitosi N. Synthesis ofα-trialkylsilyl ketones[J].Bulletin of the chemical society of Japan,1979,52(9):2646-2652.
Study on Regio- and Diastereoselective Epoxide Opening/Halogenation of Geminal Bis(silyl) 2,3-Epoxy Alcohols
HU Jia, GAO Lu*, SONG Zhen-lei*
(West China School of Pharmacy, Sichuan University, Chengdu 610041, China)
The reaction of C3-geminal bis(silane) substituted 2,3-epoxy alcohols with TsCl, Br2and I2proceeds by a regio- and diastereoselective C3-expoxide opening/halogenation process, giving 15 novel C3-halogenerated products in 64%~87% yields. The structures were characterized by1H NMR,13C NMR and HR-MS(ESI). The stereochemistry was confirmed by X-ray single crystal diffraction of 3-(dimethyl(phenyl)silyl)-3-iodo-3-(trimethylsilyl)butane-1,2-diol(8d).
geminal bis(silane); 2,3-epoxy alcohol; epoxide ring opening; halogenation; regioselectivity; diastereoselectivity; synthesis
2017-02-06
国家自然科学基金资助项目(21622202, 21502125)
胡佳(1991-),男,汉族,四川达州人,硕士研究生,主要从事天然产物及药物合成研究。 E-mail: hjleon@outlook.com
高璐,副教授, E-mail: lugao@scu.edu.cn; 宋振雷,教授, Tel. 028-85501876, E-mail: zhenleisong@scu.edu.cn
O623.42; O623.43
A
10.15952/j.cnki.cjsc.1005-1511.2017.04.17021
猜你喜欢
大电机技术(2022年5期)2022-11-17 08:14:04
云南化工(2021年11期)2022-01-12 06:06:10
世界农药(2019年4期)2019-12-30 06:25:14
电子测试(2018年1期)2018-04-18 11:52:24
材料科学与工程学报(2016年1期)2017-01-15 13:33:53
合成化学(2015年4期)2016-01-17 09:01:04
海军航空大学学报(2015年1期)2015-11-11 17:22:41
无机化学学报(2014年8期)2014-02-28 17:32:36
无机化学学报(2014年7期)2014-02-28 17:32:16
化学工业与工程(2012年6期)2012-02-10 03:21:05
