
硬皮病鉴别诊断
2017-04-06 19:30:17梁敏锐姜智星邹和建黄琼
上海医药 2017年25期
梁敏锐+姜智星+邹和建+黄琼

摘 要 硬皮病(系统性硬化症)是一种少见的多脏器受累的系统性自身免疫性疾病,以皮肤纤维化为特征表现。许多疾病临床上也可表现为皮肤变硬和组织纤维化,易与硬皮病混淆。本文强调皮肤病理对硬皮病鉴别诊断的重要性,综述了成人硬肿病、硬化性黏液水肿、嗜酸性筋膜炎、慢性移植物抗宿主病、肾源性系统性纤维化、硬化萎缩性苔藓和僵硬皮肤综合征等硬皮病类似疾病的临床表现、病因、病理形态和治疗方法。
关键词 硬皮病 系统性硬化症 鉴别诊断
中图分类号:R593.25 文献标识码:A 文章编号:1006-1533(2017)S1-0012-05
Differential Diagnosis of Scleroderma*
LIANG Minrui 1,2, JIANG Zhixing 1,2, ZOU Hejian 1,2, HUANG Qiong3**
(1. Division of Rheumatology, Huashan Hospital, Fudan University, Shanghai 200040, China; 2. Institute of Rheumatology, Immunology and Allergy, Fudan University, Shanghai 200040, China; 3. Department of Dermatology, Huashan Hospital, Fudan University, Shanghai 200040, China)
ABSTRACT Scleroderma (systemic sclerosis, SSc) is a rare systemic autoimmune disease with multiple organ manifestations, characterizing of skin fibrosis. Many conditions presenting with clinical hardening skin and tissue fibrosis can be confused with scleroderma. We emphasize the distinct morphological difference between scleroderma and other conditions. This article reviews the clinical presentation, etiology, morphology, and treatment options available for scleroderma mimics, including scleredema adultorum, scleromyxedema, eosinophilic fasciitis (EF), chronic graft-versus-host disease (cGVHD), nephrogenic systemic fibrosis (NSF), lichen sclerosus et atrophicus (LSA), stiff skin syndrome (SSS) and so on.
KEY WORDS scleroderma; systemic sclerosis (SSc); differential diagnosis
硬皮病(scleroderma)又稱为系统性硬化症(systemic sclerosis, SSc)或系统性硬皮病(systemic scleroderma),是一种系统性的自身免疫性疾病。皮肤除增厚变硬外,还可出现雷诺现象、指端溃疡、毛细血管扩张、甲翳毛细血管异常等,内脏受累则表现为间质性肺病变、肺动脉高压和胃食管反流等,常伴特异性自身抗体的出现。根据皮肤增厚变硬是否由肢端延伸至肢体近端(锁骨、肘关节、膝关节的近端),SSc可分为弥漫性SSc(diffuse cutaneous SSc, dcSSc)和局限性SSc(limited cutaneous SSc, lcSSc);CREST综合征是SSc的一个亚型,表现为钙质沉着(calcinosis, C)、雷诺现象(Raynauds syndrome, R)、食道运动功能障碍(esophageal dysmotility, E)、指端硬化(sclerodactyly, S)和毛细血管扩张(telangiectasis, T);另外1%患者无典型皮肤硬化,即无硬皮病型SSc(scleroderma sine SSc)。与SSc对应的是局灶性硬皮病(localized scleroderma, LS),后者是一种引起皮肤增厚变硬的非系统性的皮肤病变,无结构性血管损害和内脏累及。局灶性硬皮病可分为五种亚型:硬斑病、泛发性硬斑病、大疱性硬斑病、线状硬斑病和深部硬斑病[1]。此外,一些表现为皮肤硬化的疾病易与硬皮病混淆(表1),可根据病因、特征性临床表现、皮肤病理改变进行鉴别诊断,及时准确地诊断有助于判断预后和指导治疗。
1 成人硬肿病
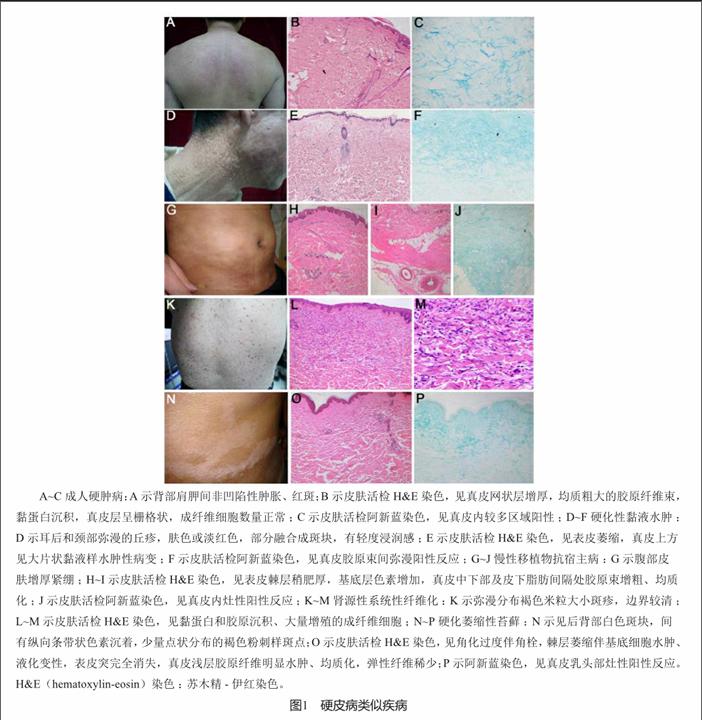
成人硬肿病(scleredema adultorum)一种病因未明的,以黏蛋白沉积、皮肤变硬为主要表现的疾病。成人硬肿病一般累及颈部、肩部、背部和腰部,特征性表现为“床垫征”(图1A),一般不累及手足[2]。偶有舌、咽和食管上段受累,从而影响张口和吞咽[3]。通常分为三型:I型为Buschke硬肿病,好发于青年,男女比1∶2,常与细菌或病毒前驱感染有关,如流感、麻疹、流行性腮腺炎、水痘或链球菌。通常为自限性,常于数月内自发缓解,然而部分患者皮肤病变可持续性或周期性加重;II型常与单克隆球蛋白血症有关,达45%的患者进展为骨髓瘤[4];III型与糖尿病有关,表现为急性起病的皮肤病变,包括一过性皮疹、荨麻疹和皮肤划痕征阳性。此外,有些硬肿病患者和其他疾病相关,不能归于以上分类,如合并胰岛素瘤、继发性甲状旁腺功能亢进、类风湿性关节炎、干燥综合征、HIV感染或接触有机溶剂等。皮疹形态和SSc类似,然而分布区域不同于SSc,一般无雷诺现象、甲翳毛细血管异常或自身抗体。如发现单克隆球蛋白血症,需警惕血液系统疾患。
组织学发现表皮层一般无异常,真皮网状层可增厚,充满了均质粗大的胶原纤维束,其间填充黏蛋白,导致真皮层呈栅格状,成纤维细胞数量正常或减少(图1B和1C)。
成人硬肿病以经验性治疗为主,临床证据有限。临床上曾尝试使用多种免疫调节药物,疗效有待证实。UVA-1或PUVA光疗效果值得期待[5]。近來有报道显示他莫昔芬和秋水仙碱可能使成人硬肿病患者获益[6]。颈肩部的理疗也很重要。当伴发的感染控制后,成人硬肿病也可在数月至数年内自发缓解。
2 硬化性黏液水肿
硬化性黏液水肿(scleromyxedema)是黏液水肿性苔藓的一种特殊类型,临床上以局限性或全身性苔藓样丘疹及硬皮病样改变,病理上特征性表现为黏蛋白沉积于真皮层,成纤维细胞增生和纤维化病变。约80%的患者存在单克隆球蛋白血症(大部分为IgG l链),然而与皮肤病变程度及内脏累及的关系暂不明确。不伴有甲状腺疾病。该病常见于50岁以上的患者,在增厚变硬的皮肤上常出现泛发的对称性的苔藓样丘疹(2~3 mm),主要累及手和腕部,随后累及肘、躯干、颈部、前额和耳后(图1D),常呈线性分布,皮肤发红明显,可有色素沉着,一般不累及手掌、头皮和黏膜。随着疾病进展,丘疹可融合成片。关节受累导致关节活动受限和关节挛缩。内脏受累不多见,但也有文献报道食管、肌肉、心脏和中枢神经系统受累,后者可表现为精神紊乱、脑病、癫痫、失语、记忆减退、抑郁和腕管综合征。
皮肤活检结果显示黏蛋白弥漫性沉积于真皮层的中到深部,胶原纤维、断裂的弹力纤维和增生的成纤维细胞分布其中。真皮浅层及血管周有淋巴细胞浸润(图1E和1F)。
硬化性黏液水肿尚无确切治疗方法,大部分免疫抑制治疗效果欠佳。个案报道丙种球蛋白输注有效。有报道自体造血干细胞移植对合并原发性淀粉样变的患者有效[7]。
3 嗜酸性筋膜炎
嗜酸性筋膜炎(eosinophilic fasciitis, EF)也称为伴有嗜酸性粒细胞增多的弥漫性筋膜炎或Shulman综合征或筋膜炎-脂膜炎综合征,临床上易与硬皮病混淆。嗜酸性筋膜炎好发于40~50岁,男女比为2∶1,典型表现为对称性的肢体和躯干变硬,较少累及面部和手指。筋膜炎症和纤维化引起皮下深静脉走行处出现皱褶,即“沟槽征”。可在创伤或外力作用后急性或亚急性起病,伴有肌无力、肌痛和关节痛,血沉和C反应蛋白升高及高丙种球蛋白血症。80%患者外周血嗜酸性粒细胞升高。由于炎症位于皮下脂肪组织深层,因而与SSc表现不同,浅层皮肤捏起后有皱褶,且不累及手指,无雷诺现象和甲翳毛细血管异常,自身抗体通常为阴性,一般无系统累及。磁共振有助于诊断嗜酸性筋膜炎,T1、T2序列见筋膜增厚,抑脂序列见筋膜强化[8]。筋膜活检是诊断金标准,磁共振引导下进行筋膜活检可提高诊断准确性[9]。尽管合并肿瘤的发生率不高,仍需仔细排查潜在的白血病、淋巴瘤、骨髓瘤或骨髓异常增生综合征等,必要时行骨髓涂片及骨髓活检。除了血液系统肿瘤外,还需警惕的是实体肿瘤在嗜酸性筋膜炎患者中的发病率也较为增高[10],如乳腺癌[11]、前列腺癌[10]和肺腺癌[10]。
该病累及筋膜和深层皮下脂肪组织,出现筋膜增厚、纤维化、硬化伴水肿,急性期常见黏液状变和纤维素样坏死。可见炎症细胞浸润,包括淋巴细胞、浆细胞、组织细胞和数目不等的嗜酸性粒细胞,有时可见含生发中心的淋巴滤泡。真皮表层一般不受累,半数患者真皮深层可见纤维化改变,伴有汗腺萎缩。表层肌肉也可累及,出现局灶性坏死和再生灶。
糖皮质激素为一线治疗,有效率达到70%[12]。初始剂量为泼尼松(或等效糖皮质激素)0.5~1.0 mg/kg/d,治疗应答后可减量,减至泼尼松20 mg/d时减量速度变缓,一般需要12~18个月达到完全应答。如病情进展迅速,出现广泛累及,明显消瘦,躯干或颈部受累时,初始可使用糖皮质激素冲击治疗,可联合使用免疫抑制剂,如甲氨蝶呤或霉酚酸酯,采用这一治疗策略有助于迅速控制病情,减少激素累积剂量。免疫抑制剂也可用于难治性和复发的患者。一项回顾了88例嗜酸性筋膜炎病例的综述中指出,不良预后因素(定义为难治性或经延长治疗仍存留有纤维化病变)包括年轻时起病,合并硬斑病和躯干受累[13]。如通过以上治疗仍无应答,应尽早排查潜在的恶性病变。理疗有助于改善关节挛缩和活动障碍。总体而言预后较好,大部分患者可达到完全缓解并治愈。
4 慢性移植物抗宿主病
约70%的患者接受同种异体骨髓移植后出现慢性移植物抗宿主病(chronic graft-versus-host disease, cGVHD),系可累及皮肤及内脏的系统性病变。cGVHD的皮肤病变表现多样,约15%的患者可出现硬皮病样改变[14],即硬化型cGVHD(图1G)。临床表现取决于皮肤累及深度,累及真皮乳头层可导致皮肤变薄、萎缩性白色丘疹和斑块;累及真皮网状层可导致硬化斑块,往往固定,常累及躯干和肢体。挛缩可累及关节,表皮萎缩可导致破溃形成,往往见于腿部和胫前;累及脂肪层和皮下组织往往隐匿起病,出现皮下坚硬的结节,呈“橘皮样”外观,可伴皮肤色素沉着。深层组织受累需要与嗜酸性筋膜炎鉴别,后者可有“沟槽征”和深部木样硬化。皮肤组织活检可见表皮萎缩、角化过度、毛囊角栓,凋亡的角质形成细胞,浅层胶原均质化,如病变加深可见增粗的胶原束和附属器减少。皮下组织和筋膜累及时表现为相应部位的增厚、水肿和纤维化(图1H、1I和1J),伴不同程度的淋巴细胞、组织细胞和嗜酸性粒细胞浸润。
与SSc不同的是,cGVHD主要累及躯干和四肢(而非从双手受累起始),皮损分布不均,cGVHD与局灶性硬皮病较难鉴别,然而黏膜受累和骨髓移植病史更支持cGVHD的诊断。
可采用光疗来治疗硬化型cGVHD,尤其是UVA和UVA-1[15]。然而其有效性尚未通过临床试验证实。系统性免疫抑制治疗的效果不确切。两项随机试验采用系统性糖皮质激素和霉酚酸酯用于治疗硬化型cGVHD,结果显示疗效不佳[15]。回顾性研究指出雷帕霉素靶蛋白抑制剂(如依维莫司或西罗莫司)可使75%的硬化型cGVHD患者获益[15]。小型病例队列研究提示伊马替尼对硬化型cGVHD有效,相关临床试验也在开展中[16-17]。
5 肾源性系统性纤维化
最初在1997年于美国圣地亚哥发现第一例肾源性系统性纤维化(nephrogenic systemic fibrosis, NSF)患者,并于2000年发表于《柳叶刀》杂志[18]。目前认为该病发生于肾功能不全患者,主要是慢性肾脏病(chronic kidney disease, CKD)-5期患者,主要危險因素为使用含钆造影剂,一项队列研究指出在使用造影剂钆的CKD-5期患者中,13%出现NSF[19]。钆对NSF的直接致病性有待进一步证实,但美国食品药品监督管理局已于2006年12月建议中重度肾脏病患者避免使用含钆造影剂。虽然多数患者长期透析,但本病与肾替代治疗的类型或路径无相关性,10%的患者未曾行透析。该病无地域或人种差异,男女比为1∶1,发病年龄跨度大(8~87岁),有儿童病例报道[20]。主要表现为快速进展的皮肤纤维化结节,并融合成片,呈木样硬化,可出现棕黄色蜡样斑(图1K),伴有烧灼感和严重的瘙痒,主要累及肢体远端(膝关节及肘关节的远端),躯干也可受累,但面部不受累。NSF可导致严重的关节挛缩及致命的内脏病变。与SSc不同的是,NSF无雷诺现象、毛细血管扩张、甲翳微循环异常或肢端骨质溶解,也不出现SSc特异性抗体。皮肤活检可见成纤维细胞增生,黏蛋白和胶原沉积,但较少炎症细胞浸润(图1L和1M)。目前无特效治疗手段,糖皮质激素、环孢素、组胺受体拮抗剂和沙利度胺通常无效,而血浆置换、光疗和西罗莫司部分有效,伊马替尼对治疗NSF具有一定的前景[21]。
6 硬化萎缩性苔藓
硬化萎缩性苔藓(lichen sclerosus et atrophicus, LSA)是一种少见的慢性炎症性皮肤病变,常表现为生殖器萎缩性斑块,其他部位亦可受累(图1N)。目前病因不明,推测与遗传易感性、自身免疫、螺旋体感染等密切相关。硬斑病同样可以出现萎缩性斑块,与螺旋体感染和自身免疫也密切相关。两者也可同时出现,临床表现和病理改变有所重合,因而也有研究认为两种疾病可归为同一疾病谱[22]。然而与硬斑病不同的是硬化萎缩性苔藓瘙痒更为明显[23],硬斑病特征性表现为网状内皮的硬化,胶原纤维肿胀,血管周炎症细胞浸润及附属器的丢失。而硬化萎缩性苔藓表现为毛囊栓塞和真皮乳头层苔藓样浸润及胶原均质化(图1O和1P)。有研究认为真皮浅层是否存在弹力纤维有助于两者的鉴别,弹力纤维在硬斑病中存在,而在硬化萎缩性苔藓中消失[23]。皮肤镜对两者的鉴别可提供诊断线索,硬化性萎缩性苔藓可见粉刺样开口和白斑,分别与毛囊栓塞和萎缩性病变对应,而硬斑病在皮肤镜下可见纤维化条带。两者治疗方法有相同之处,包括外用糖皮质激素、抗疟药、秋水仙碱、甲氨蝶呤和光疗。研究显示UVA和窄谱UVB对硬化萎缩性苔藓的疗效优于硬斑病[24]。对于病变进展迅速的患者,可考虑甲氨蝶呤和糖皮质激素联合使用。
7 僵硬皮肤综合征
僵硬皮肤综合征(stiff skin syndrome, SSS)又称为先天性筋膜发育不良(congenital facial dystrophy),1977年由Esterly和McKusik首先报道[25]。有报道家族遗传的SSS病例,为常染色体显性遗传,研究发现原纤蛋白-1 (FBN1) 基因突变,从而使FBN1和细胞外基质间的相互作用发生异常所致[26]。患者出生时或婴幼儿起出现皮肤发硬,逐渐进展呈岩石样坚硬,皮肤外观可正常,也可轻度多毛或色素沉着,皮损以臀部和大腿最为明显。可出现关节病变,常累及大关节,导致关节挛缩、脊柱侧弯、踮足步态或胸廓缩小,甚至引起限制性通气功能障碍和生长发育迟滞。通常无内脏或骨骼肌肉受累,无自身抗体或血管病变,无尿黏多糖排泄增加。皮肤病理可见黏蛋白沉积,成纤维细胞增生,胶原纤维粗大增多和筋膜增厚。SSS需要与SSc进行鉴别,SSS的皮损主要分布于筋膜较多部位,如环绕骨盆的臀部及大腿,以及环绕肩部的上臂,而SSc皮肤病变主要分布于双手、面部、颈肩和背部,SSS的皮损较不均匀,甚至出现团块状,但SSc的皮肤病变则更为均匀弥漫,关节挛缩在SSS中较常出现,而在SSc中少见。起病方式两者也有区别,SSS患者出生时或婴幼儿时起病,SSc则可成年起病。两者的皮肤病理特点有重合:SSS中可见黏蛋白沉积,胶原纤维粗大,胶原纤维间隙缩小,筋膜增厚,无炎症细胞浸润或血管病变;SSc的胶原纤维的形态大致正常,无黏蛋白沉积,且无筋膜累及,可有炎症细胞浸润和血管病变。SSS通常无雷诺现象、甲翳毛细血管异常和自身抗体出现[27]。SSS对糖皮质激素治疗效果不佳,以对症支持治疗和理疗为主。近来有报道霉酚
酸酯和自体造血干细胞移植对SSS可能有效[28-29],基于 FBN1靶点的治疗具有前景,有待进一步研究。
此外,另有一些硬皮病类似疾病表现为皮肤增厚变硬,如POEMS综合征、迟发型皮肤卟啉病、嗜酸性粒细胞增多-肌痛综合征、Werner综合征、苯丙酮尿症和毒油综合征等(表1),临床上可通过病史采集、体格检查和组织病理学分析进行鉴别诊断。及时准确的诊断有助于判断预后和指导治疗。总体而言,这类疾病发病机制不明,治疗手段缺乏临床试验的数据支持,治疗上具有挑战性。
参考文献
[1] Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma)[J]. Mayo Clin Proc, 1995, 70(11): 1068-1076.
[2] Meguerditchian C, Jacquet P, Beliard S, et al. Scleredema adultorum of Buschke: an under recognized skin complication of diabetes[J]. Diabetes Metab, 2006, 32(5 Pt 1): 481-484.
[3] Wright RA, Bernie H. Scleredema adultorum of Buschke with upper esophageal involvement[J]. Am J Gastroenterol, 1982, 77(1): 9-11.
[4] Dziadzio M, Anastassiades CP, Hawkins PN, et al. From scleredema to AL amyloidosis: disease progression or coincidence? Review of the literature[J]. Clin Rheumatol, 2006, 25(1): 3-15.
[5] Konemann S, Hesselmann S, Bolling T, et al. Radiotherapy of benign diseasesscleredema adultorum Buschke[J]. Strahlenther Onkol, 2004, 180(12): 811-814.
[6] Alsaeedi SH, Lee P. Treatment of scleredema diabeticorum with tamoxifen[J]. J Rheumatol, 2010, 37(12): 2636-2637.
[7] Lacy MQ, Hogan WJ, Gertz MA, et al. Successful treatment of scleromyxedema with autologous peripheral blood stem cell transplantation[J]. Arch Dermatol, 2005, 141(10): 1277-1282.
[8] Baumann F, Bruhlmann P, Andreisek G, et al. MRI for diagnosis and monitoring of patients with eosinophilic fasciitis[J]. AJR Am J Roentgenol, 2005, 184(1): 169-174.
[9] Imai T, Saitoh M, Matsumoto H. Eosinophilic fasciitis: MRI evaluation[J]. Neurology, 2003, 61(3): 416.
[10] Naschitz JE, Yeshurun D, Zuckerman E, et al. Cancer-associated fasciitis panniculitis[J]. Cancer, 1994, 73(1): 231-235.
[11] Watts RA, Merry P. Familial eosinophilic fasciitis and breast cancer[J]. Br J Rheumatol, 1994, 33(1): 93-94.
[12] Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after -what do we really know? Report of 11 patients and review of the literature[J]. Dermatology, 2006, 213(2): 93-101.
[13] Endo Y, Tamura A, Matsushima Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome[J]. Clin Rheumatol, 2007, 26(9): 1445-1451.
[14] Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. Pathogenesis and clinical manifestations of graft-versus-host disease[J]. J Am Acad Dermatol, 2012, 66(4): 515 e511-518; quiz 533-514.
[15] Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part II. Management of cutaneous graft-versus-host disease[J]. J Am Acad Dermatol,
2012, 66(4): 535 e531-516; quiz 551-532.
[16] Olivieri A, Locatelli F, Zecca M, et al. Imatinib for refractory chronic graftversus-host disease with fibrotic features[J]. Blood, 2009, 114(3): 709-718.
[17] Chen GL, Arai S, Flowers ME, et al. A phase 1 study of imatinib for corticosteroid-dependent/refractory chronic graft-versus-host disease: response does not correlate with anti-PDGFRA antibodies[J]. Blood, 2011, 118(15): 4070-4078.
[18] Cowper SE, Robin HS, Steinberg SM, et al. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients[J]. Lancet, 2000, 356(9234): 1000-1001.
[19] Todd DJ, Kagan A, Chibnik LB, et al. Cutaneous changes of nephrogenic systemic fibrosis: predictor of early mortality and association with gadolinium exposure[J]. Arthritis Rheum, 2007, 56(10): 3433-3441.
[20] Jan F, Segal JM, Dyer J, et al. Nephrogenic fibrosing dermopathy: two pediatric cases[J]. J Pediatr, 2003, 143(5): 678-681.
[21] Bernstein EJ, Schmidt-Lauber C, Kay J. Nephrogenic systemic fibrosis: a systemic fibrosing disease resulting from gadolinium exposure[J]. Best Pract Res Clin Rheumatol, 2012, 26(4): 489-503.
[22] Virdi SK, Kanwar AJ. Generalized morphea, lichen sclerosis et atrophicus associated with oral submucosal fibrosis in an adult male[J]. Indian J Dermatol Venereol Leprol, 2009, 75(1): 56-59.
[23] Kim DH, Lee KR, Kim TY, et al. Coexistence of lichen sclerosus with morphoea showing bilateral symmetry[J]. Clin Exp Dermatol, 2009, 34(7): e416-e418.
[24] Brownell I, Soter NA, Franks AG, Jr. Familial linear scleroderma (en coup de sabre) responsive to antimalarials and narrowband ultraviolet B therapy[J]. Dermatol Online J, 2007, 13(1): 11.
[25] Esterly NB. The stiff skin syndrome[J]. Birth Defects Orig Artic Ser, 1971, 7(8): 306-308.
[26] Loeys BL, Gerber EE, Riegert-Johnson D, et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome[J]. Sci Transl Med, 2010, 2(23): 23ra20.
[27] Liu T, McCalmont TH, Frieden IJ, et al. The stiff skin syndrome: case series, differential diagnosis of the stiff skin phenotype, and review of the literature[J]. Arch Dermatol, 2008, 144(10): 1351-1359.
[28] Kurtzman DJ, Wright NA, Patel M, et al. Segmental stiff skin syndrome (SSS): Two additional cases with a positive response to mycophenolate mofetil and physical therapy[J]. J Am Acad Dermatol, 2016, 75(6): e237-e239.
[29] Bandinelli F, Saccardi R, Salvadorini G, et al. Stiff skin syndrome and myeloma successfully treated with autologous haematopoietic stem cell transplantation(HSCT)[J]. Clin Exp Rheumatol, 2013, 31(2 Suppl 76): 181-183.
[30] Boin F, Hummers LK. Scleroderma-like fibrosing disorders[J]. Rheum Dis Clin North Am, 2008, 34(1): 199-220; ix.
猜你喜欢
广州中医药大学学报(2022年2期)2022-02-13 06:37:40
人人健康(2020年17期)2020-11-13 08:22:12
家庭百事通·健康一点通(2018年7期)2018-08-10 06:55:34
益寿宝典(2018年22期)2018-01-26 15:28:08
中国实用医药(2016年28期)2016-12-07 22:23:14
中国实用医药(2016年27期)2016-11-30 12:00:46
中国实用医药(2016年27期)2016-11-30 11:28:09
中外医学研究(2016年28期)2016-11-28 11:29:26
医学信息(2016年29期)2016-11-28 09:07:46
中国实用医药(2016年24期)2016-10-17 03:42:53
