
超高效液相色谱–串联质谱法测定饮用水中微囊藻毒素
2017-02-16 08:18邹康兵向彩红董玉莲
化学分析计量 2017年1期
邹康兵,向彩红,董玉莲
(国家城市供水水质监测网广州监测站,广州市自来水公司水质部,广州 510160)
超高效液相色谱–串联质谱法测定饮用水中微囊藻毒素
邹康兵,向彩红,董玉莲
(国家城市供水水质监测网广州监测站,广州市自来水公司水质部,广州 510160)
建立饮用水中微囊藻毒素(MC–RR,MC–LR)的超高效液相色谱–串联质谱检测方法。样品经PVDF针式过滤头过滤后直接进样,采用喷雾正离子源(ESI+)和多重反应监测模式(MRM)测定。MC–RR的质量浓度在0.02~10.00 μg/L范围内与色谱峰面积呈良好的线性,线性相关系数r2=0.998 9,检出限为0.096 μg/L,测定结果的相对标准偏差为6.6%~9.1%(n=7),加标回收率为99.0%~103.0%。MC–LR的质量浓度在0.1~20 μg/L范围内与色谱峰面积呈良好的线性,线性相关系数r2=0.999 2,检出限为0.188 μg/L,测定结果的相对标准偏差为4.3%~10.0%(n=7),加标回收率为93.0%~114.0%。该方法灵敏度高、重现性好,可用于饮用水中微囊藻毒素的检测。
超高效液相色谱–串联质谱法;饮用水;微囊藻毒素
淡水水体中蓝藻毒素已成为全球性的问题,世界各地经常发生蓝藻毒素中毒事件[1],我国太湖曾于2007年爆发过蓝藻严重污染事件,造成无锡自来水全城污染。许多饮用水源受蓝藻污染时,从其中检测出了微囊藻毒素(MC),因此MC引起了人们的高度关注。MC是一类环状七肽化合物,相对分子质量在1 000左右,其中Adda结构为3-氨基-9-甲氧基-10-苯基-2,6,8-三甲基-4,6-二烯酸,肽环上的2,4位,即R1和R2为两种可变的L-氨基酸,由于R1和R2的不同及3,7位上氨基酸的甲基化或去甲基化,形成了多种不同的微囊藻毒素异构体。目前已从不同微囊藻菌株中分离、鉴定出了70多种微囊藻毒素[2–4],其中毒性较大的两种藻毒素为MC–RR和MC–LR[5](R代表精氨酸,L代表亮氨酸)。MC化学性质稳定[2],具强亲水性和高耐热性,自然环境下降解非常缓慢,即使加热煮沸也无法将其去除,现行的自来水常规处理工艺如混凝、沉淀、过滤、加氯等均不能有效将其去除[6]。MC具有强大的危害性,属肝毒素,具有强致癌性,若皮肤接触或长期饮用含MC的水,可对人体造成伤害,甚至引起肝癌[7]。因此为确保饮用水安全,GB 5749–2006《生活饮用水卫生标准》将饮用水中MC–LR的限值规定为0.001 mg/L。
目前MC的检测方法主要有生物毒理检测法、酶联免疫检测法[8]和高效液相色谱法等[9–13]。生物毒理检测法不能区分毒素的异构体且定量不够准确;酶联免疫检测法不能对毒素进行良好的鉴别,有时会出现假阳性反应;高效液相色谱法灵敏度不高,而天然水体中的MC含量很低,故水样必须通过固相萃取柱富集浓缩才能定量分析。笔者用超高效液相色谱串联质谱法对MC进行测定,样品经针式过滤器过滤后直接进样,免去了样品富集步骤,减少了样品和有机溶剂的用量,同时消除了共提取物的干扰,提高了检测灵敏度。
1 实验部分
1.1 主要仪器与试剂
超高效液相色谱–串联质谱仪:UPLC/Quattro Premier XE MS型,配电喷雾离子源(ESI),美国Waters公司;
MC–LR,MC–RR固体标准品:HPLC级,50µg,纯度大于95℅,–25℃保存,台湾T.A.S公司;
乙腈:HPLC级,纯度不低于99.9℅,德国Merck公司;
甲酸:HPLC级,纯度不低于98.0℅,中国上海CNW公司;
PVDF针式过滤头:30 mm×0.22 µm,广州洁特生物制品有限公司;
实验用水为纯水(Watsons 屈臣氏蒸馏水)。
1.2 标准溶液配制
1.2.1 标准储备溶液
MC–RR标准储备溶液:25 mg/L,称取适量MC–RR标准品,用移液器加入2 mL水–甲醇(体积比为50∶50,下同),充分摇匀,密封,4℃避光保存,待用。
MC–LR标准储备溶液:25 mg/L,称取适量MC–LR标准品,用移液器加入2 mL水–甲醇,充分摇匀,密封,4℃避光保存,待用。
1.2.2 标准使用溶液
MC–RR标准使用液:20.00 µg/L,用移液器取0.04 mL MC–RR标准储备溶液于盛有水–甲醇的50 mL棕色容量瓶中,再用水–甲醇稀释至标线,密封,4℃避光保存。
MC–LR标准使用液:100.00 µg/L,用移液器取0.20 mL MC–LR标准储备溶液于盛有水–甲醇的50 mL棕色容量瓶中,再用水–甲醇稀释至标线,密封,4℃避光保存。
1.3 标准曲线绘制
MC–RR:准确吸取MC–RR标准使用液0.01,0.05,0.25,0.50,1.0,20.0,2.50,5.00 mL分别置于10.0 mL容量瓶中,用水–甲醇定容,摇匀。制得质量浓度分别为0.02,0.10,0.50,1.00,2.00,4.00,5.00,10.00 µg/L的MC–RR系列标准工作溶液,分别测定,绘制标准工作曲线。
MC–LR:准确吸取MC–LR标准使用液0.01,0.05 ,0.25,0.50,1.00,2.00 mL于10.0 mL容量瓶中,用水–甲醇定容,摇匀,制得质量浓度分别为0.10,0.50,2.50,5.00,10.00,20.00 µg/L的MC–LR系列标准工作溶液,分别测定,绘制标准工作曲线。
1.4 仪器工作条件
1.4.1 液相色谱仪
色谱柱:Acquity UPLC BEH shield RP18柱(50 mm×2.1 mm,1.7 µm,美国Waters公司);柱温:30℃;流动相:0.1%甲酸水溶液–乙腈(90:10);流量:0.30 mL/min;梯度洗脱;进样体积:10 µL。
1.4.2 质谱仪
离子源:ESI+模式;毛细管电压:2.5 kV;锥孔电压:MC–LR为65 V,MC–RR为50 V;脱溶剂气流量:500 L/h;气帘气流量:50 L/h;检测方式:多反应检测(MRM)模式。
1.5 样品预处理方法
分析前,将样品溶液于4℃避光保存;分析时,用PVDF针式过滤头将样品溶液过滤。
1.6 定性及定量离子
MC–RR:选择m/z439.8作为定性离子,m/z134.8作为定量离子。
MC–LR:选择m/z375.3作为定性离子,m/z135.3作为定量离子。
1.7 结果计算
根据测得水样中微囊藻毒素的色谱峰面积(或色谱峰高),从校准曲线上直接查得微囊藻毒素的浓度。水样中微囊藻毒素的含量按式(1)计算:

式中:c——水样中微囊藻毒素的质量浓度,µg/L;
cE——标样中微囊藻毒素的质量浓度,µg/L;
AE——测得微囊藻毒素标样的色谱峰面积;
A——测得水样中微囊藻毒素的色谱峰面积。
2 结果与讨论
2.1 样品处理方法优化
由于超高效液相色谱–串联质谱法灵敏度高,直接进样检出限均能达到国标限值10倍以下,水样不需要再富集,可直接进样分析。对PTFE,PVDF,MCE 3种不同材质的针式过滤器的加标回收结果进行比较,在3种不同针式过滤器中随机各抽取7个针式过滤器,用纯水做加标试验,MC–RR的加标浓度为1,9 µg/L,MC–LR的加标浓度为2 ,18 µg/L。试验结果表明,MCE对MC–RR和MC–LR的加标回收率分别为91.4%~101.0%,93.5%~102.7%;PVDF对MC–RR和MC–LR的加标回收率分别为88.0%~100.9%,88.0%~101.7%;PTFE针式过滤器对于MC–LR和MC–RR的加标回收率均在80%左右。可见MCE和PVDF两种滤膜的针式过滤器比较满意,对MC–LR和MC–RR分析时不会带来新的干扰,以及不会产生吸附,对测定结果的准确度没有影响,且价格相当,因此均可以用来过滤样品,本实验选择PVDF针式过滤器。
2.2 液相色谱条件的优化
2.2.1 流动相及洗脱方式
由于MC易溶于水、甲醇和乙腈,因此选择水–甲醇及水–乙腈作为流动相进行比较试验。为使待测物能有效分离,并获最高灵敏度,选择等度和梯度两种洗脱方式对以下4种流动相进行比较试验[14–15]。(1) 0.1%甲酸水溶液–乙腈(50∶50);(2) 0.1%甲酸水溶液–乙腈(70∶30);(3) 0.1%甲酸水溶液–乙腈(90∶10);(4) 0.1%甲酸水溶液–甲醇(90∶10)。试验结果表明,等度洗脱时洗脱效果不理想,梯度洗脱时流动相(3)峰形好,响应值高,出峰时间快,在2 min内可较好地分离微囊藻毒素,因此选择流动相为0.1%甲酸水溶液–乙腈(90:10)。
2.2.2 流动相中甲酸含量
为增加电离效果在流动相中加入少量甲酸,并对加入甲酸的含量进行优化。对以下3种甲酸含量的流动相进行比较试验:(1) 0.2%甲酸水溶液–乙腈(体积比90∶10);(2) 0.1%甲酸水溶液–乙腈(体积比90∶10);(3)水–乙腈(体积比90∶10)。采用梯度洗脱方式,对比3种流动相在保留时间、响应强度方面的差异。
试验结果表明,加入甲酸后离子化效率明显增高,使用0.1%甲酸水溶液–乙腈(90∶10),出峰时间快,响应强度高,且峰形好。因此采用0.1%甲酸水溶液–乙腈(90∶10)为流动相,梯度洗脱。在2 min内分离MC–RR和MC–LR,保留时间分别为1.43,1.77 min,见图1、图2。流动相梯度洗脱程序见表1。

图1 MC–LR标准样品色谱图
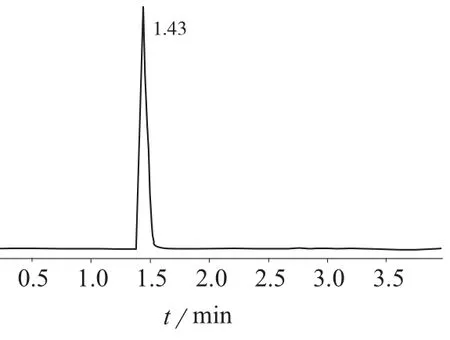
图2 MC–RR标准样品色谱图
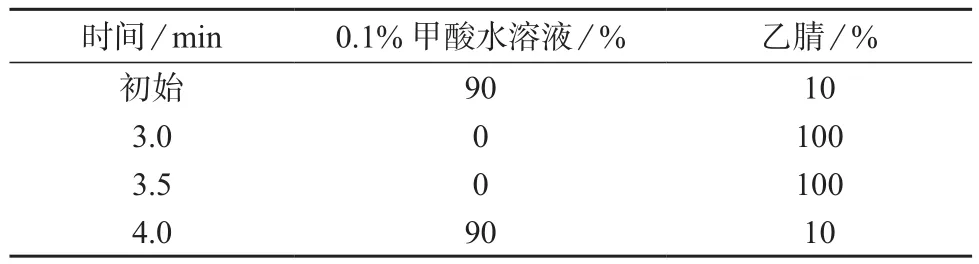
表1 流动相梯度洗脱程序
2.3 质谱条件优化
MC相对分子质量大、极性强,适合ESI+电离模式,将MC–RR,MC–LR标准品分别配制成10 µg/L 的标准溶液,通过全扫描方式确定其母离子,MC–RR的母离子[M+2H]2+的存在形式最高,故选择519.98(RR[M+2H]2+)为母离子;MC–LR以[M+H]+的形式为主,选择996.0(LR[M+H]+)为母离子。
MC–RR和MC–LR的碎片离子中均存在碎片m/z135且丰度最高,因此选择其作为定量离子,其中MC–RR为m/z134.8,MC–LR为m/z135.3;选择丰度稍小的作为定性离子,MC–RR为m/z439.8,MC–LR为m/z375.3。经试验确定的其它条件如锥孔电压、气帘气流量等见1.4.2。
2.4 配制标准溶液时溶剂比例的优化
实验中发现配制标准溶液时溶剂对MC–RR和MC–LR的响应值有较大影响,对水、甲醇的体积比为100∶0,80∶20,60∶40,50∶50,40∶60,20∶80,100∶0时进行比较试验。结果表明,用纯水配制时响应值最低,随着加入甲醇比例的提高,响应值逐渐增大,当甲醇超过一定比例后,响应值又开始降低。甲醇与水的体积比为50∶50时响应值最理想。同时做回收试验时发现,采取先配制成水–甲醇(50∶50)的合成水样后,再加标,然后过滤,上机检测,回收率较好。
2.5 工作曲线方程与检出限
在选择的仪器工作条件下,对1.3中系列标准工作溶液进行测定,以待测组分的色谱峰面积Y为纵坐标,以相应的质量浓度X为横坐标,绘制标准曲线。在超纯水中加入MC–RR,MC–LR的质量浓度分别为0.02,0.10 µg/L,重复测定7次,计算测定结果的标准偏差,按MDL=3.14s计算方法检出限。MC–RR,MC–LR的线性方程和检出限见表2。由表2可知,MC–RR和MC–LR均具有良好的线性,线性相关系数均在0.995以上。MC–RR和MC–LR的检出限分别为0.096,0.188 µg/L。
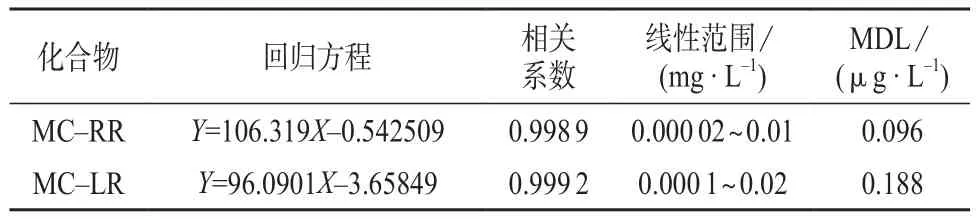
表2 回归方程、相关系数和方法检出限
2.6 精密度与回收试验
分别对纯水、自来水、水源水水样进行测定,未检出MC–RR和MC–LR,然后进行加标回收试验,重复测定7次,结果见表3。由表3可知,MC–RR测定结果的相对标准偏差在6.6%~9.1%之间,平均回收率为99.0%~103.0%;MC–LR测定结果的相对标准偏差在4.3%~10.0%之间,平均回收率为93.0%~114.0%。可见回收率均在80%~120%之间,满足GB 5749–2006《 生活饮用水标准检验方法水质分析质量控制》对测量准确度的要求。
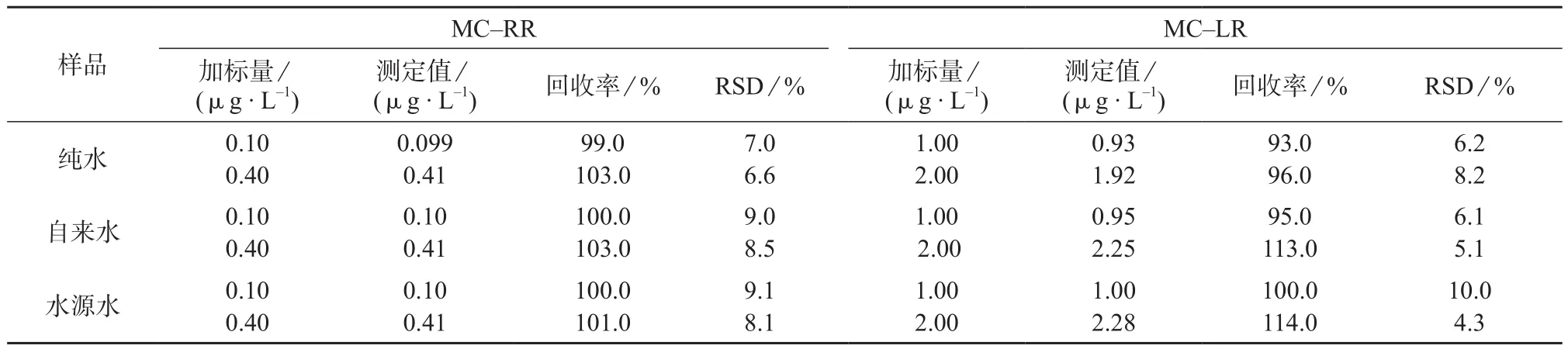
表3 精密度和回收试验结果(n=7)
3 结语
采用超高效液相色谱串联质谱法对饮用水中微囊藻毒素MC–RR,MC–LR进行测定,与国标方法相比,该方法简化了样品的前处理步骤,样品不需要经过固相萃取富集或衍生化处理,经过针式过滤器过滤直接进样分析,缩短了分析时间,且分析过程中有机试剂用量少。该方法灵敏度高,精密度、准确度满足现行国内及国际水质检测标准的要求。
[1] 谢平.蓝藻水华及其次生危害[J].水生态学杂志,2015,36(4): 1–13.
[2] Kunimitsu K. Chemistry and toxicology of cylic hepatpetide toxins,the microcystins from cyanobacteria[J]. Cult Coll 1994,105–33.
[3] Xiuyuan Zhang,Kuo He ,Ruiping Zhao,et al. Cloning scfv from hybridomas using a rational strategy: Application as a receptor to sensitive detection microcystin-LR in water[J]. Chemosphere,2016,160: 230–236.
[4] Fangfang Wang,Shuzhen Liu,Mingxia Lin,et al. Colorimetric detection of microcystin-LR based on disassembly of orientaggregated gold nanoparticle dimmers[J]. Biosensors and Bioelectronics,2015,68: 475–480.
[5] Caroline Murphy,Edwina Stack,Svetlana Krivelo,et al. Detection of the cyanobacterial toxin,microcystin-LR,using a novel recombinant antibody-based optical-planar waveguide platform[J]. Biosensors and Bioelectronics,2015,67: 708–714.
[6] 谢平.水生动物体内的微囊藻毒素及其对人类健康的潜在威胁[M].北京:科学出版社,2006.
[7] 谢平.微囊藻毒素对人类健康影响相关研究的回顾[J].湖泊科学,2009,21(5): 603–613.
[8] Hou Li,Ding Yunhua,Zhang Lili,et al. An ultrasensitive competitive immunosensor for impedimetric detection of microcystin-LR via antibody-conjugated enzymatic biocatalytic precipitation[J]. Sensors and Actuators B,2016,233: 63–70.
[9] Zhang Lifeng,Ping Xiaofei,Yang Zhaoguang. Determination of microcystin-LR in surface water using high-performance liquid chromatography /tandem electrospray ionization mass detector[J]. Talanta,2004,62: 193–200.
[10] Monica Barco,Cintia Flores,Josep Rivera et al. Determination of microcystin variants and related peptides present in a water bloom of Planktothrix(Oscillatoria)rubescens in a Spanish drinking water reservoir by LC/ESI–MS[J]. Toxicon,2004,44: 881–886.
[11] Cong Liming,Huang Baifen,Chen Qi,et al. Determination of trace amount of microcystins in water samples using liquid chromatography coupled with triple quadrupole mass spectrometry[J]. Analytica Chimica Acta,2006,569: 157–168.
[12] Ma Jiping,Yan Fengli,Chen Fengxi,et al. C18–Functionalized magnetic silica nanoparticles for solid phase extraction of microcystin-LR in reservoir water samples followed by HPLC–DAD determination[J]. Journal of Liquid Chromatography&Related Technologies, 2015,38: 655–661.
[13] Li Lianli,Liu Guoting,Wu Yuqing,et al. Determination of microcystin-LR in enxironmental water by magnetic solid phase extraction-high performance liquid chromatography[J]. Chinese Journal of Analytical Chemistry,2015,43(12): 1 876–1 881.
[14] 王静,庞晓露,刘铮铮,等.超高效液相色谱/串联质谱法分析水中的微囊藻毒素[J].色谱,2006,24 (4): 335–338.
[15] Arthur Zastepa,Frances R Pick,Jules M Blais,et al. Analysis of intracellular and extracellular microcystin variants in sediments and pore waters by accelerated solvent extraction and high performance liquid chromatography-tandem mass spectrometry[J]. Analytica Chimica Acta,2015,872: 26–34.
赛默飞中国生物制品开发实验室正式揭幕助力中国生物制药事业快速升级
不久前,赛默飞世尔科技公司(以下简称:赛默飞)的中国生物制品开发实验室正式揭幕,该实验室致力于帮助用户提高创新及拓展生产能力。新实验室落户于赛默飞上海中国创新中心,总投资额超过200万美元,配有总面积约50平方米的细胞培养实验室以及近70平方米的分析实验室。
“十三五”规划中明确指出医药研发是未来重点推进任务,旨在促进技术创新和产业发展,将重点发展重大疾病化学药物、生物技术药物、新疫苗等多个创新药物品类;此外,还对整个生物医药产业提出了新的发展目标,即进一步促进产业的国际化发展,大力推动本土企业实现药品质量标准和体系与国际接轨。依托其全球资源和在生物制药领域的领先技术和服务能力,赛默飞中国生物制品开发实验室旨在为生物制品业的客户提供一流的技术、服务和管理,帮助他们延展培养基和工艺方面的生产和创新能力,并能够助力其满足和符合日益严格的生产和审批标准。具体而言,该实验室将帮助客户提高研发成功率,加快其研发速度,提供配方研发、小批量试产及大规模量产供货等一系列服务。立足中国并熟谙市场的研发团队将根据中国客户的实际需求,在新药研发、临床试验、药物生产、进入市场效率等各个环节都能够为客户提供有力帮助和支持。
( 中国分析计量网)
海洋局携国标委、质检总局全面部署“十三五”海洋标准
不久前,国家海洋局召集各海洋部门及相关单位,在北京共聚一堂,共商海洋标准计量质量改革发展大计。这也是首次在海洋领域召开的全国海洋质量管理工作会议,对推进海洋标准计量质量工作、落实建设海洋强国战略具有重要的现实意义。
近年来,国家海洋局全面加强海洋标准计量质量工作,大力推进改革创新,着力提升能力水平。“十二五”以来,国家海洋局发布了海洋观测预报及防灾减灾等7个标准体系,正在制定海岛保护与利用等5个标准体系,海洋标准体系已初步建立。发布了32项海洋国家标准和108项海洋行业标准,近3年每年海洋标准项目立项近百项。
在海洋计量工作方面,目前已有41套海洋计量标准装置涵盖海流等13个要素,其中“十二五”期间新增7套,有33套社会公用计量标准成为统一全国海洋计量单位制的最高标准。14套海洋校准装置通过了实验室认可,大幅拓展了校准服务的能力范围。成立了全国海洋专用计量器具计量技术委员会,发布和正在制定15项国家计量技术规范,填补了海洋领域空白。海洋社会公用计量标准和标准物质基本能够覆盖海洋水文、气象和化学领域80%以上的仪器种类,年均提供检定校准服务5 000余台套,海洋计量检测服务能力处于国内先进行列,保障了全国海洋仪器设备的量值准确可靠。
据介绍,目前我国海洋质量管理体系已初步建立。通过《海洋观测预报条例》等法律文件,强化了海洋质量管理工作,加强了海洋公益服务的质量管理。初步建立了海洋领域质量监督组织体系,已有62家海洋检验检测机构通过了国家级计量认证评审,新增7家海洋单位通过了ISO 9000质量管理体系认证。同时,推进了海洋专项和海洋公益服务的质量工作,提升了重点海洋产品的质量。
据国家海洋局副局长林山青介绍,今年该局先后分别与国家标准委、质检总局联合印发了《全国海洋标准化“十三五”发展规划》和《全国海洋计量“十三五”发展规划》。“十三五”期间,该局将在更高起点上推动海洋标准计量质量工作大发展:(1)着力提高海洋标准有效供给,大力实施“海洋标准化+”工程,积极开展海洋标准“走出去”工程,提高标准化创新能力;(2)着力提高海洋计量检测水平,加强海洋公益服务的计量检测工作,强化海洋产品的计量检测服务,提升海洋计量国际化水平;(3)着力加强和规范海洋质量管理,大力推进质量管理体系建设,创新质量管理手段和方式,提升海洋产品质量。
( 中国分析计量网)
Determination of Microcystin in Drinking Water by UPLC–MS–MS
Zou Kangbing, Xiang Caihong, Dong Yulian
(Guangzhou Monitoring Station, National Water Quality Monitoring Net of City Water Supply,Water Quality Department of Guangzhou Water Supply Co., Guangzhou 510160, China)
A method was established for the determination of microcystin (MC–RR, MC–LR) in drinking water by high performance liquid chromatography tandem mass spectrometry. The sample was fltered by PVDF needle type flter,then sampled directly and detected with electrospray positive ion source(ESI+) and multiple reaction monitoring mode (MRM). The concentration of MC–RR was linear with chromatographic peak area in the range of 0.02–10.00 μg/L with the correlation coeffcient (r2) of 0.998 9,the detection limit was 0.096 μg/L,the relative standard deviation of detection results was 6.6%–9.1%(n=7),and the addition recoveries was 99.0%–103.0%. The concentration of MC–LR was linear with chromatographic peak area in the range of 0.1–20 μg/L with the correlation coeffcient (r2) of 0.999 2, the detection limit was 0.188 μg/L,the relative standard deviation of detection results was 4.3%–10.0% (n=7),and the addition recoveries was 93.0%–114.0%. The method has higher sensitivity and good reproducibility,it is suitable for the determination of microcystin (MC–RR, MC–LR) in drinking water.
high performance liquid chromatography tandem mass spectrometry; drinking water; microcystin
O657.7
:A
:1008–6145(2017)01–0042–05
10.3969/j.issn.1008–6145.2017.01.010
联系人:邹康兵;E-mail: 79411581@qq.com
2016–11–12
猜你喜欢
煤化工(2022年3期)2022-07-08
核化学与放射化学(2022年2期)2022-04-28
色谱(2021年7期)2021-06-07
江苏农业学报(2019年1期)2019-09-10
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
中南大学学报(自然科学版)(2016年2期)2017-01-19
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
