
磁性多壁碳纳米管固相萃取/高效液相色谱-串联质谱法测定蜂蜜中多组分兽药残留
2017-02-14 09:19徐潇颖罗金文陈万勤赵超群
分析测试学报 2017年1期
徐潇颖,罗金文,陈万勤,赵超群,刘 柱
(浙江省食品药品检验研究院,浙江 杭州 310052)
磁性多壁碳纳米管固相萃取/高效液相色谱-串联质谱法测定蜂蜜中多组分兽药残留
徐潇颖,罗金文,陈万勤,赵超群,刘 柱*
(浙江省食品药品检验研究院,浙江 杭州 310052)
建立了以磁性多壁碳纳米管为吸附剂的分散固相萃取/高效液相色谱-串联质谱检测方法,对蜂蜜中3大类44种兽药残留进行测定。样品经pH 4.0的Na2EDTA-Mcllvaine缓冲液提取,加入自制磁性多壁碳纳米管吸附目标物。目标物经10%氨水-甲醇洗脱后,液相色谱-串联质谱MRM模式进行定性定量分析。44种药物在1~40 ng/mL浓度范围内线性关系良好,相关系数均大于0.99;在3个不同浓度添加水平下,回收率为78.0%~105.1%,相对标准偏差(RSD)为1.2%~8.9%,检出限为0.2~2.0 μg/kg。结果表明,该方法简单方便,易于操作,为蜂蜜中磺胺类、喹诺酮类以及硝基咪唑类兽药残留的测定提供了新途径。
磁性多壁碳纳米管;磺胺;硝基咪唑;喹诺酮;蜂蜜
磺胺类、喹诺酮类、硝基咪唑类药物均为抗菌药,在养殖业中应用广泛[1]。在目前蜂养殖业中,蜜蜂易得细菌性疾病,蜂农为预防蜂病的发生,往往会给蜜蜂喂食抗菌类药物,致使蜂蜜中兽药残留问题屡禁不止。2002年,欧盟以抗生素残留超标为由,禁止中国蜂蜜出口,虽然随后解除了该项禁止,但对蜂蜜中兽药残留的检测工作仍持续开展。由于蜂蜜中糖含量较高,直接测定痕量的兽药残留存在一定困难,检测通常以固相萃取小柱净化来减少杂质干扰。目前市场上商品化的固相萃取小柱(MWCNTs)是一种具有高稳定性和强吸附性能的纳米材料,比表面积大,一般由2~50层石墨片组成,直径在几到几十纳米之间,长度可达几十甚至上百微米[2-6],对有机物、金属离子和有机金属化合物等具有较强的富集能力[7]。近年来,MWCNTs因具有类似C18的吸附性能,被作为一种新型的固相萃取剂,广泛用于农药残留[8-16]和兽药残留[17-18]的检测。对磁性颗粒进行表面修饰,不仅能高效地捕集样品基质中的痕量目标分析物,而且很好地克服了传统固相萃取小柱在样品处理过程中易堵塞和操作繁琐等问题,使分离和富集过程变得简捷、快速和高效。Kumar等[19]将其用于氧氟沙星的富集分析研究,Zhao等[20]在食用油中多环芳烃的分析检测也使用了磁性MWCNTs。本文利用实验室研制的磁性MWCNTs净化蜂蜜样品,并对净化过程中的主要影响因素进行优化,采用超高效液相色谱-串联质谱技术同时测定了蜂蜜中3大类44种兽药残留。该方法简单方便,易于操作,从而为蜂蜜中磺胺类、喹诺酮类以及硝基咪唑类兽药残留的测定提供了支持。
1 实验部分
1.1 仪器与试剂
Agilent 6460液相色谱-串联质谱仪;Milli-Q超纯水器(美国Millipore公司);氮气吹干仪(Biotage公司);涡旋混合器(上海琪特公司);酸度计(Mettler Toledo公司)。
甲醇、乙腈(色谱纯,德国Merck公司);甲酸(色谱纯,美国Fluka公司);乙二胺四乙酸二钠、磷酸二氢钠、磷酸氢二钠、氨水、氯化铁、氯化亚铁、浓硝酸(分析纯,国药集团化学试剂有限公司);MWCNTs纯度>97%,直径20~40 nm,长度>5 μm(深圳市纳米港有限公司)。
实验所用44种兽药的标准物质均购自Dr.Ehrenstorfer GmbH。
1.2 标准溶液配制
分别准确称取10 mg上述标准品置于10 mL容量瓶中,用甲醇定容,配成浓度为1 mg/mL的储备液,密封储存于-18 ℃冰箱中,分别移取20 μL 1 mg/mL的储备液于10 mL容量瓶中,用甲醇定容后配制成2 μg/mL的混合标准溶液。使用前稀释成一系列浓度的标准溶液。
1.3 磁性多壁碳纳米管的制备
1.3.1 MWCNTs羧化称取1.5 g多壁碳纳米管粉末置于250 mL单口烧瓶中,加入150 mL浓硝酸,超声振荡1 h,移入恒温水浴槽中,于60 ℃回流12 h,冷却后,除去上层酸液,反复水洗至中性,于100 ℃下烘干4 h。
1.3.2 磁 化称取0.36 g羧化MWCNTs粉末置于250 mL双口烧瓶中,加入225 mL水,超声20 min后加入0.18 g无水氯化铁,移入恒温水浴槽中于60 ℃在氮气保护下振摇30 min,加入0.45 g氯化亚铁后继续振摇30 min,加入150 mL 25%氨水振摇2 h后,取出,冷却,除去上层液体,水洗至中性,放入电热鼓风干燥箱在50 ℃下烘干24 h。MWCNTs磁化后可被磁铁吸附,未磁化和磁化后多壁碳纳米管的电镜照片见图1。

1.4 样品前处理方法
称取蜂蜜样品2 g于250 mL烧杯中,加入100 mg磁性MWCNTs,加200 mL pH 4.0的Na2EDTA-Mcllvaine溶液,超声5 min,使磁性MWCNTs均匀分布于提取液中,提取60 min后,用磁铁对磁性MWCNTs进行吸附,用吸管吸取清水对磁性MWCNTs进行淋洗后去除洗液,加入10 mL 10%氨水-甲醇后超声5 min,收集洗脱液,重复洗脱1次,合并两次洗脱液,于40 ℃下氮吹近干,用初始比例流动相定容至1.0 mL,过0.22 μm有机滤膜,待测。
1.5 色谱和质谱条件
1.5.1 色谱条件色谱柱:Thermo C18(150 mm×2.1 mm,2.6 μm)或相同效能色谱柱;流速:0.2 mL/min;柱温:35 ℃;进样量:5 μL;流动相为乙腈(A)-0.1%甲酸水溶液(B),梯度洗脱程序:0~4 min,90% B;4~8 min,90%~65% B;8~12 min,65%~55% B;12~14 min,55%~70% B;14~16 min,70%~90% B;16~18 min,90% B。
1.5.2 质谱条件电喷雾离子源(ESI);正离子扫描模式;多反应监测(MRM)模式;源电压4 kV;毛细管温度400 ℃;鞘气流速12 L/min;兽药化合物的质谱参数见表1。

表1 MRM监测模式下44种药物的质谱条件
*quantitation ion
2 结果与讨论
2.1 提取溶液的优化
蜂蜜样品中含有大量糖分,通常用水稀释后进行提取,在对蜂蜜样品进行加水稀释后的磁性吸附实验中,易出现喹诺酮类药物回收率低。主要是由于不同类抗生素药物的pKa值范围较宽的缘故。实验分别采用pH 4.0的Na2EDTA-Mcllvaine溶液、pH 6.0和pH 8.0的磷酸盐缓冲液以及0.1 mol/L氢氧化钠溶液(pH 12.0)作为提取溶液进行试验。结果表明,使用酸性提取液的提取效果优于碱性提取液,其中经pH 4.0的Na2EDTA-Mcllvaine溶液稀释后进行磁性碳纳米管吸附实验得到的回收率较高,且基质干扰较小,故选择其作为提取溶液。
2.2 磁性MWCNTs加入量的优化
在相同样品提取液中,磁性多壁碳纳米管的加入量对样品中多组分兽药残留的回收率也存在直接影响,分别称取20,60,100,150 mg的MWCNTs加入100 mL样品提取液中,当加标量为20 μg/kg时,磺胺类、喹诺酮类、硝基咪唑类药物的回收率随着MWCNTs加入量的增加而增加,当加入量大于100 mg时回收率变化趋于平缓。因此,当样品称取量为2 g时,磁性MWCNTs的最佳加入量为100 mg。
2.3 洗脱条件的优化
分别采用甲醇、5%甲酸-甲醇及5%氨水-甲醇作为洗脱液,对吸附目标物的磁性MWCNTs进行洗脱。结果显示,目标物在碱性条件下的洗脱效果优于甲醇和5%甲酸-甲醇的洗脱效果。单独采用甲醇作为洗脱溶剂时,喹诺酮类药物的回收率偏低,主要是因为酸性提取液使磺胺类、喹诺酮类及硝基咪唑类药物不同程度结合了质子,在甲醇中添加氨水能中和目标物结合的质子,使目标物呈中性,更利于洗脱,提高其回收率。该现象与文献[21-22]的研究结果相一致。在碱性洗脱条件下,进一步对不同体积分数的氨水-甲醇(5%,10%,15%)进行比较,结果表明随着洗脱液中氨水比例增大,洗脱后的样液颜色变深,杂质干扰变大,当洗脱液中氨水比例由10%升至15%时,3类药物的回收率增加不明显。因此,选择10 mL 10%氨水-甲醇分两次进行洗脱。
2.4 色谱条件的优化
兽药多残留检测依赖于色谱柱的选择,不同色谱柱对不同类别兽药的分离效果存在一定差异,实验分别考察了单独测定磺胺类、喹诺酮类、硝基咪唑类兽药残留时常用的Thermo C18(150 mm×2.1 mm,2.6 μm),Waters Atlantis®T3(150 mm×2.1 mm,5.0 μm),ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm)的分离效果。结果显示,Thermo C18(150 mm×2.1 mm,2.6 μm)在MRM模式下对44种组分可实现较好的分离,且各色谱峰峰形较好,杂质干扰较小。因此,本实验选择Thermo C18(150 mm×2.1 mm,2.6 μm)对待测物进行分离。
对比了甲醇和乙腈作为流动相的有机相时的分离效果,发现乙腈作为强极性洗脱溶剂,可提高待分离组分的离子化效率,而在水相中加入0.1%甲酸时,各化合物在ESI+模式下的离子化效果更好,峰形有所改善,灵敏度也相应提高,因此本实验选取乙腈及0.1%甲酸水作为流动相。分四通道对44种化合物的MRM色谱图进行提取,结果如图2所示。
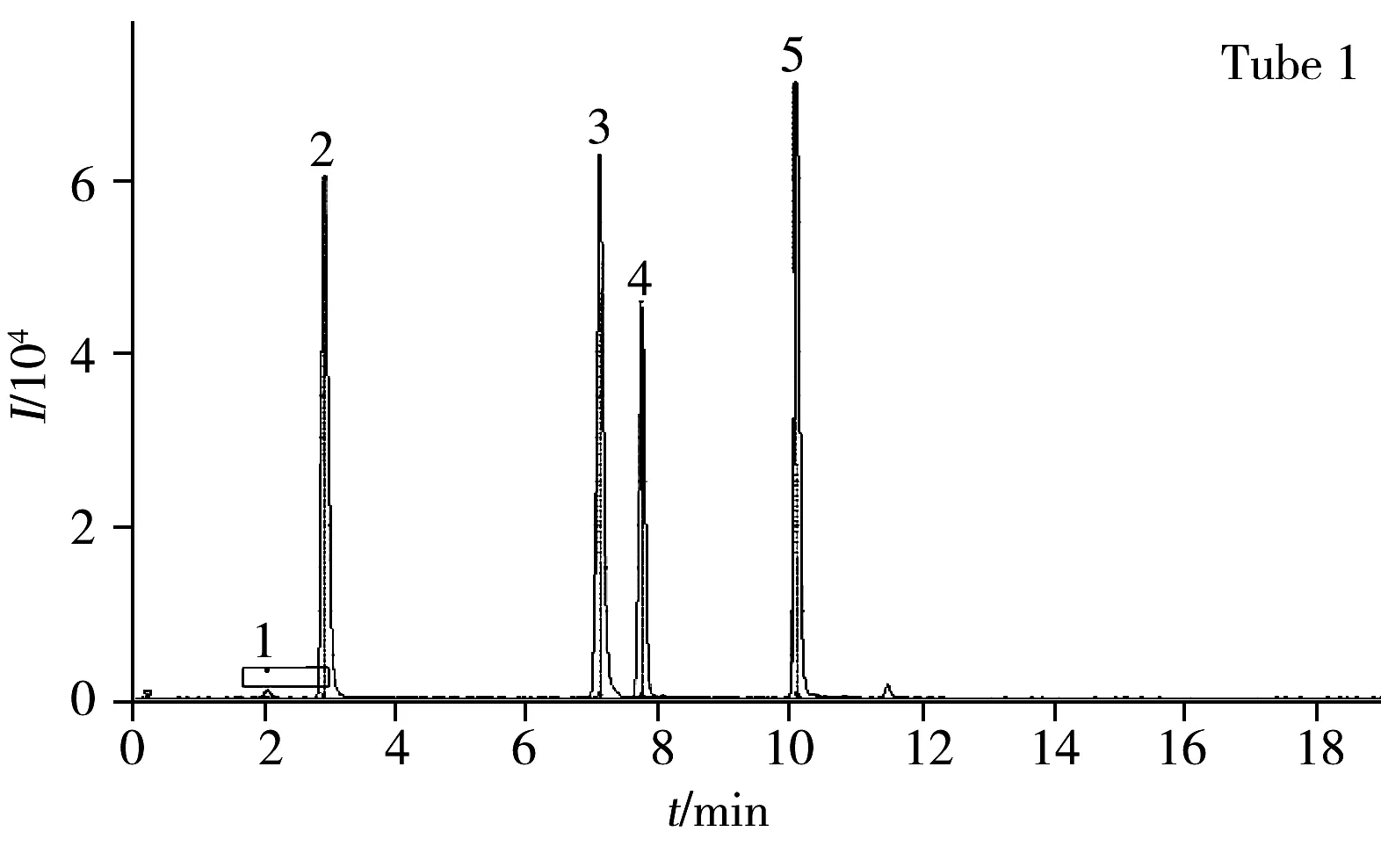

2.5 线性关系、检出限与加标回收率
采用空白基质溶液配制系列浓度的标准溶液,以各目标物的峰面积(Y)为纵坐标,质量浓度(X,ng/mL)为横坐标进行线性拟合。结果表明,3类药物均在1~40 ng/mL范围内呈良好的线性关系,相关系数(r2)大于0.99。对阴性蜂蜜样品进行加标实验,添加5,10,30 μg/kg 3个浓度水平的混合标准溶液,每个浓度设4个平行,按“1.4”方法进行样品前处理,回收率、相对标准偏差及检出限见表2。20种磺胺类、17种喹诺酮类及7种硝基咪唑类药物的回收率分别为83.6%~105.1%,78.0%~101.1%及87.7%~99.5%;检出限为0.2~2.0 μg/kg。磁化后的碳纳米管,与曹慧等[21]采用多壁碳纳米管进行蜂蜜中兽残测定,所得的磺胺类、喹诺酮类及硝基咪唑类药物回收率相符,说明磁化处理对碳纳米管的富集作用无影响。侯建波等[23]采用HLB固相萃取柱进行前处理净化后,测得磺胺类、喹诺酮类及硝基咪唑类药物的回收率分别为52.5%~114.0%,62.8%~110.0%及67.5%~111.0%,说明本文采用磁性碳纳米管对这3类药物的回收率可达到HLB固相萃取柱净化处理的回收水平。

表2 44种兽药的回收率测定结果及方法检出限(n=12)
2.6 实际蜂蜜样品的分析
应用本方法对96批蜂蜜样品进行测定,其中2批蜂蜜样品中分别检出5.6 μg/kg环丙沙星和7.0 μg/kg诺氟沙星。与各药物的现行国标方法GB/T 23412-2009的测定结果(环丙沙星和诺氟沙星含量分别为6.7,6.5 μg/kg)进行比对,未发现假阴性情况。
3 结 论
与传统的前处理过程中采用固相萃取小柱净化相比,磁性MWCNTs在进行样品前处理过程中可均匀分布于提取液中,对目标物的吸附更彻底,待吸附完成后,利用磁性吸引,使吸附目标物的磁性MWCNTs快速被收集,磁化后的MWCNTs将磁性分离技术和固相吸附剂结合,有效解决了吸附剂难回收问题,大大简化了前处理过程,达到了多类兽残同时提取,且净化效果理想,价格低。利用自制的磁性MWCNTs固相萃取材料测定蜂蜜中20种磺胺类、17种喹诺酮类及7种硝基咪唑类兽药残留,回收率为78.0%~105.1%,相对标准偏差为1.2%~8.9%。其中,磺胺类、喹诺酮类及硝基咪唑类的平均回收率分别为88.1%,88.2%,91.5%,与采用固相萃取柱进行前处理后测定的3类兽残的回收率相当。实验结果初步显示出磁性MWCNTs在前处理净化过程中的省时、节约、便捷等特点,这为蜂蜜中多组分兽药残留的测定提供了新方法。
[1] He Q,Kong X Q,Li J H,Le A S,Wu S M.Chin.J.Anal.Lab.(何强,孔祥虹,李建华,乐爱山,吴双民.分析试验室),2010,29(8):61-65.
[2] Huang Y,Yuan Y L,Zhou Z D.Appl.SurfaceSci.,2014,292:378-386.
[3] Pistone A,Lannazzo D,Fazio M.PhysiacaB-CondensedMatter,2014,434:88-91.
[4] Demir A,Baykal A,Sozeri H.Synth.Met.,2014,187:75-80.
[5] Yin M,Wang M L,Miao F.Carbon,2012,50(6):2162-2170.
[6] Majidi M R,Salimi A,Alipour E.J.Chin.Chem.Soc.,2013,60(12):1473-1478.
[7] Katsumata H,Matsumoto T,Kaneco S.Microchem.J.,2008,88(1):82-86.
[8] Zhou Q,Xiao J,Wang W.Talanta,2006,68(4):1309-1315.
[9] Guo L,Lee H K.J.Chromatogr.A,2011,1218(52):9321-9327.
[10] Liu X Y,Zhang H X,Liu M C.J.Chromatogr.A,2008,1212(1/2):10-15.
[11] Wang W D,Huang Y M,Shu W Q.J.Chromatogr.A,2007,1173(1/2):27-36.
[12] Liu X Y,Ji Y S,Zhang Y H.J.Chromatogr.A,2007,1165(1/2):10-17.
[13] Zhou Q X,Wang W D,Xiao J P.Anal.Chim.Acta,2006,559(2):200-206.
[14] Zhou Q X,Wang W D,Ding Y J.Chin.Chem.Lett.,2008,19(1):95-98.
[15] Dong M F,Ma Y Q,Zhao E C.Microchim.Acta,2009,165(1/2):123-128.
[16] Rong J F,Wei H,Huang H S,Li Y J,Xu M Z.J.Instrum.Anal.(荣杰峰,韦航,黄伙水,李亦军,许美珠.分析测试学报),2016,35(1):8-15
[17] Zhao H X,Liu H P,Yan Z Y.Chin.J.Chromatogr.(赵海香,刘海萍,闫早婴.色谱),2014,32(3):294-298.[18] Xu Y,Chen H Y,Zhao Q.FoodChem.,2013,140(1/2):83-90.
[19] Kumar D R,Manoj D,Santhanalakshmi J.J.Nanosci.Nanotechnol.,2014,13:1-11.
[20] Zhao Q,Wei F,Luo Y B.J.Agric.FoodChem.,2011,59(24):12794-12800.
[21] Cao H,Chen X Z,Zhu Y,Li Z G.Chem.J.Chin.Univ.(曹慧,陈小珍,朱岩,李祖光.高等学校化学学报),2013,12(34):2710-2715.
[22] Cao H,Chen X Z,Zhu Y,Li Z G.NewCarbonMater.(曹慧,陈小珍,朱岩,李祖光.新型碳材料),2015,30(6):572-578.
[23] Hou J B,Xie W,Chen X M,Xi J Y,Qian Y.Chin.J.Chromatogr.(侯建波,谢文,陈笑梅,奚君阳,钱艳.色谱),2011,29(6):535-542.
Determination of Veterinary Drugs Residues in Honey by High Performance Liquid Chromatography-Tandem Mass Spectrometry with Magnetic Multi-walled Carbon Nanotubes
XU Xiao-ying,LUO Jin-wen,CHEN Wan-qin,ZHAO Chao-qun,LIU Zhu*
(Zhejiang Institute for Food and Drug Control,Hangzhou 310052,China)
A method using magnetic multi-walled carbon nanotubes(mMWCNTs) as the absorbent followed by ultra-performance liquid chromatography-tandem mass spectrometry was established for the determination of 44 veterinary drugs residues in honey.Analytes were extracted with Na2EDTA-Mcllvaine buffer solution(pH 4.0) before self-made mMWCNTs were added to absorb veterinary drugs.After extraction with mMWCNTs and followed by elution with 10% ammonia-methanol,target analytes were analyzed using multiple reaction monitoring(MRM) mode.The correlation coefficients of linear calibration curves were over 0.99 in the concentration range of 1-40 ng/mL.The recoveries of 44 analytes at three spiked concentrations ranged from 78.0% to 105.1%,with relative standard deviations(RSDs) of 1.2%-8.9%.The limits of detection(LODs,S/N≥3) were 0.2-2.0 μg/kg.With the advantages of simplicity,sensitivity and good precision, this method provides a new way for the determination of multi-veterinary drug residues in honey.
magnetic multi-walled carbon nanotubes;sulfonamide;nitroimidazole;quinolone;honey
10.3969/j.issn.1004-4957.2017.01.010
2016-07-26;
2016-08-29
浙江省科技计划项目(2015C37054);浙江省科技厅重大项目(2014C02001)
*通讯作者:刘 柱,硕士,中级工程师,研究方向:食品安全与分析技术,Tel:0571-87180322,E-mail:zliu82@126.com
O657.63;TQ460.72
A
1004-4957(2017)01-0061-06
猜你喜欢
农产品加工(2022年14期)2022-11-17
中学生学习报(2021年20期)2021-11-24
鞍钢技术(2021年3期)2021-06-11
世界最新医学信息文摘(2021年12期)2021-06-09
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
浙江大学学报(工学版)(2016年9期)2016-06-05
中国卫生标准管理(2015年25期)2016-01-14
原子与分子物理学报(2015年3期)2015-11-24
郑州大学学报(理学版)(2014年4期)2014-03-01
