
丁酸氯维地平降解杂质的合成
2017-02-07 08:59:06褚小琴闫少杰李树军黄汉忠王平保
合成化学 2017年1期
褚小琴, 闫少杰, 李树军, 张 远, 黄汉忠*, 王平保*
(1. 天津医科大学 研究生院,天津 300070; 2. 天津药物研究院有限公司 化学制药部,天津 300193)
·制药技术·
丁酸氯维地平降解杂质的合成
褚小琴1,2, 闫少杰2, 李树军2, 张 远2, 黄汉忠2*, 王平保2*
(1. 天津医科大学 研究生院,天津 300070; 2. 天津药物研究院有限公司 化学制药部,天津 300193)
以4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3,5-吡啶二羧酸(2-氰基乙基)(甲基)酯(5)为起始原料,合成了丁酸氯维地平的5种降解杂质:4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3,5-吡啶二羧酸单甲酯(A), 4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3-吡啶羧酸甲酯(B), 4-(2,3-二氯苯基)-2,6-二甲基-3,5-吡啶二羧酸单甲酯(C), 4-(2,3-二氯苯基)-2,6-二甲基-3,5-吡啶二羧酸(丁酰氧基甲基)(甲基)酯(D)和4-(2,3-二氯苯基)-2,6-二甲基-3-吡啶羧酸甲酯(E)。其中A由5水解制得;B由A脱羧制得;C由5氧化后再经水解制得;D由C和丁酸氯甲酯缩合制得;E由C脱羧制得,化合物结构经1H NMR和MS(ESI)确证。
丁酸氯维地平; 降解杂质; 药物合成
丁酸氯维地平,商品名为Cleviprex,化学名为4-(2,3-二氯苯基)-2,6-二甲基-1,4-二氢吡啶-3,5-二羧酸(丁酰氧基甲基)(甲基)酯,是由英国AstraZeneca公司研发的一种超短效二氢吡啶类钙通道拮抗剂[1]。2008年8月由美国FDA批准上市,主要用于包括急性高血压的治疗以及心脏手术、经皮冠状动脉介入治疗等手术后的血压控制[2],同时它作为注射用乳剂上市适用于不适宜口服或口服无效的高血压治疗。
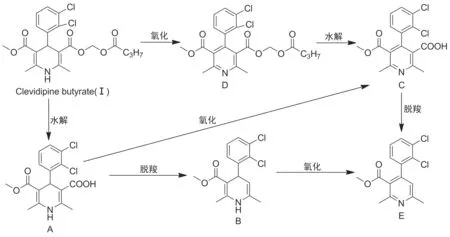
Scheme 1
降解杂质的研究是新药开发的重要组成环节,不但可以为原料药和制剂的工艺质量控制提供依据,也有利于提高产品质量标准的可靠性。丁酸氯维地平是典型的二氢吡啶类药物,其降解杂质的产生可能有以下几个原因:(1)吡啶环上1,4-H极不稳定,在光照或其他氧化条件下,容易发生脱氢芳构化;(2) 3-位和5-位的酯键容易断裂而水解,3-位酯基在酸性或碱性的水环境中更容易水解成酸;(3) 水解产物在高温条件下易发生脱羧继续降解。因此可推测丁酸氯维地平的原料药和制剂在生产和储藏过程中容易发生Scheme 1所示的反应,形成一系列降解杂质。
目前,有关丁酸氯维地平的合成研究较多,但对其降解杂质的合成报道相对较少。杂质4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3,5-吡啶二羧酸单甲酯(A)作为一种制备丁酸氯维地平的重要中间体,其合成报道较多。其合成主要通过Jung等[3]报道的碱性条件下水解丙腈酯的方法,但收率较低。宋志勇等[4]以杂质A为起始原料,在N,N-二甲基甲酰胺中回流脱羧制得杂质4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3-吡啶羧酸甲酯(B);杂质B经Jones试剂氧化制得杂质4-(2,3-二氯苯基)-2,6-二甲基-3-吡啶羧酸甲酯(E);丁酸氯维地平经过氧化尿素氧化制得杂质4-(2,3-二氯苯基)-2,6-二甲基-3,5-吡啶二羧酸(丁酰氧基甲基)(甲基)酯(D)。该路线中杂质D和杂质E采用分次氧化,不仅增加了氧化剂的种类,不利于控制成本,而且增加了副产物的种类,需用柱色谱进行分离纯化,操作复杂。杂质4-(2,3-二氯苯基)-2,6-二甲基-3,5-吡啶二羧酸单甲酯(C)的合成方法目前还未见报道,林秀云等[5]研究发现,在长期光照试验下,原料药主成分易氧化为杂质C,因此对其合成路线研究同样具有重要意义。
鉴于此,本文开发一种原料经济易得、操作简便、收率及纯度均较高合成杂质A~E的方法。参照文献[6-9]方法合成丁酸氯维地平,以2,3-二氯苯甲醛(1)与乙酰乙酸甲酯(2)经Knoevenagel缩合反应制得2,3-二氯亚苄基乙酰乙酸甲酯(3); 3与3-氨基丁烯酸丙腈酯(4)经Michael加成、脱水环合制得4-(2,3-二氯苯基)-1,4-二氢-2,6-二甲基-3,5-吡啶二羧酸(2-氰基乙基)(甲基)酯(5); 5经氢氧化钾水解制得4-(2, 3-二氯苯基)-1,4-二氢-2,6-二甲基-3,5-吡啶二羧酸单甲酯钾盐(6);最后,6与正丁酸氯甲酯反应合成丁酸氯维地平。
本文以5为起始原料,合成降解杂质A~E。 A由5水解制得;B由A脱羧制得;5氧化得中间体7,再经水解制得C; D由C和丁酸氯甲酯缩合制得;E由C脱羧制得,合成路线见Scheme 2,化合物结构经1H NMR和MS(ESI)确证。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent 6520型Accurate-Mass-Q-TOF/MS型质谱仪;Agilent 1200型高效液相色谱仪。
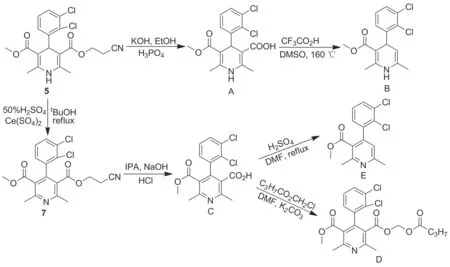
Scheme 2
5参照文献[6-9]方法合成,纯度≥99%(HPLC);其余所用试剂均为分析纯。
1.2 合成
(1) A的合成
在反应瓶中加入5 8.00 g(19.6 mmol),氢氧化钾1.02 g(25.5 mmol)和无水乙醇70 mL,于25 ℃搅拌使其溶解;反应5 h。抽滤,滤饼用无水乙醇(10 mL)洗涤,干燥得淡黄色固体;加至80 mL纯水中,用40%稀磷酸调至pH 2,于室温搅拌1.5 h。抽滤,滤饼干燥得淡黄色固体A 5.58 g,收率80.09%,纯度98.50%;1H NMR(MeOD)δ: 2.26(s, 6H, CH3), 3.55(s, 3H, CH3), 5.42(s, 1H, CH), 7.11(t,J=7.8 Hz, 1H, ArH), 7.24(d,J=9.6 Hz, 1H, ArH), 7.31(d,J=9.6 Hz, 1H, ArH); MS(ESI)m/z: 356.04{[M+H]+}。
(2) B的合成
在反应瓶中加入A 4.50 g(12.6 mmol)和二甲基亚砜45 mL,氮气气氛下搅拌使其溶解;缓慢滴加三氟乙酸10 mL,滴毕,升温至160 ℃,反应1 h。冷却至25 ℃,减压浓缩,剩余物加入纯水50 mL,用碳酸钠固体调至pH 7,用二氯甲烷(3×50 mL)萃取,合并萃取液,依次用饱和食盐水(45 mL)洗涤,无水硫酸镁干燥,蒸干得白色粗品,经硅胶柱层析[洗脱剂:V(石油醚)∶V(乙酸乙酯)=3 ∶1)]纯化得白色固体B 3.15 g,收率79.87%,纯度98.71%;1H NMRδ: 1.69(s, 3H, CH3), 2.37(s, 3H, CH3), 3.46(s, 3H, CH3), 4.72(d,J=4.8 Hz, 1H, CH), 5.03(d,J=4.8 Hz, 1H, CH), 5.13(s, 1H, NH), 7.11(t,J=2.4 Hz, 1H, ArH), 7.22(d,J=2.8 Hz, 1H, ArH), 7.23(d,J=3.2 Hz, 1H, ArH); MS(ESI)m/z: 312.04 {[M+H]+}。
(3) C的合成
在反应瓶中依次加入5 20.00 g(24.4 mmol)和叔丁醇200 mL,搅拌使其均匀;加入50%硫酸50 mL,于25 ℃滴加20%Ce(SO4)2溶液120 mL,滴毕,回流反应1 h。自然冷却至25 ℃,滴加52%氢氧化钠溶液至pH 7,减压浓缩,残余物加水200 mL,于室温搅拌1 h。过滤,滤饼干燥得淡黄色固体7 7.96 g。
将上述固体7加入异丙醇80 mL,于25 ℃搅拌使其溶解;于15 ℃加入氢氧化钠0.93 g (23.4 mmol),反应6 h。减压浓缩,残余物加水60 mL,用1 mol·L-1盐酸调至pH 1,搅拌1 h。抽滤,滤饼干燥得淡黄色固体C 6.05 g,收率34.93%,纯度98.23%;1H NMR(DMSO-d6)δ: 2.48~2.56(s, 6H, CH3), 3.48(s, 3H, CH3), 7.16(d,J=8.8 Hz, 1H, ArH), 7.39(t,J=7.8 Hz, 1H, ArH), 7.66(d,J=9.6 Hz, 1H, ArH), 13.32(s, 1H, COOH); MS(ESI)m/z: 354.02{[M+H]+}。
(4) D的合成
在反应瓶中加入C 5.00 g(14.1 mmol),碳酸钾2.53 g(18.33 mmol)和N,N-二甲基甲酰胺50 mL,搅拌下于25 ℃滴加正丁酸氯甲酯2.51 mL(19.74 mmol),滴毕,升温至85 ℃,反应6.5 h。冷却至25 ℃,过滤除去碳酸钾,滤液减压浓缩,剩余物加水50 mL,用乙酸乙酯萃取(3×50 mL),合并萃取液,依次用饱和食盐水(45 mL)洗涤,无水硫酸钠干燥,减压浓缩至干,用异丙醚重结晶得白色固体D 3.20 g,收率49.90%,纯度98.94%;1H NMRδ: 0.91(t,J=4.9 Hz, 3H, CH3), 1.57~1.62(m, 2H, CH2), 2.23(t,J=7.6 Hz, 2H, CH2), 2.61(s, 3H, CH3), 2.63(s, 3H, CH3), 3.51(s, 3H, CH3), 5.64(q,J=8.4 Hz , 2H, CH2), 7.04(d,J=9.2 Hz, 1H, ArH), 7.20(t,J=8.0 Hz, 1H, ArH), 7.44(d,J=9.2 Hz, 1H, ArH); MS(ESI)m/z: 454.08 {[M+H]+}。
(5) E的合成
在反应瓶中加入C 5.00 g(14.1 mmol), 2 mol·L-1硫酸10 mL和N,N-二甲基甲酰胺50 mL,搅拌下回流反应5 h。冷却至25 ℃,减压蒸干,残留物加入水60 mL,用52%氢氧化钠溶液调至pH 7,用乙酸乙酯(3×60 mL)萃取,合并有机相,依次用饱和食盐水(55 mL)洗涤,无水硫酸钠干燥,减压蒸干,用正己烷重结晶得淡黄色粉末E 1.75 g,收率39.96%,纯度99.43%;1H NMRδ: 2.55(s, 3H, CH3), 2.62(s, 3H, CH3), 3.53(s, 3H, CH3), 6.90(s, 1H, CH), 7.06(d,J=9.2 Hz, 1H, ArH), 7.21(q,J=8.1 Hz, 1H, ArH), 7.46(d,J=9.6 Hz, 1H, ArH); MS(ESI)m/z: 310.03{[M+H]+}。
探讨了丁酸氯维地平原料药或制剂中可能产生的五种降解杂质A 、B、 C、 D、 E的形成原因,明确了在丁酸氯维地平原料药或制剂的生产或储存环境中,要注意尽量避免高湿,高温和强光照射,减少产品中降解杂质的产生,对产品质量、安全性及有效性均有重要的指导意义。
以中间体5为起始原料,经水解、氧化及脱羧等反应合成了A、 B、 C、 D、 E五种杂质。
(1) 杂质A采用改变溶媒(无水乙醇代替50%乙醇),改变碱的种类(氢氧化钾代替氢氧化钠),较文献[3]方法提高了水解产物的收率(由52.40%提高至80.09%);杂质B采用二甲基亚砜作溶媒,在160 ℃条件下,加三氟乙酸脱羧,较文献[4]直接在N,N-二甲基甲酰胺回流下脱羧,缩短了反应时间,纯度可达到98.5%以上。
(2) 设计了杂质C的合成路线,采用化合物5氧化形成芳构化中间体7,再水解的方法,避免了因形成过氧酸杂质而难以分离纯化的缺点;以50%硫酸和20%Ce(SO4)2为氧化剂,芳构化完全,且产物容易分离,纯度较高。
(3) 杂质D、 E在杂质C的基础上分别经缩合和脱羧得到,杂质D较文献[4]方法简化了工艺流程,无需使用额外的氧化剂,节约合成成本;杂质E较文献[4]方法改进了后处理与纯化方式,用正己烷重结晶代替柱色谱分离,简化了操作步骤。
所有合成的杂质均通过1H NMR和MS(ESI)确证了其结构,且经HPLC归一化法测得纯度均在98%以上,可作为质量研究的杂质对照品,用于方法学验证,提高丁酸氯维地平原料药及制剂质量标准的可靠性和准确性。
[1] 李灵,毕开顺,胡欣. 丁酸氯维地平注射乳的药理及临床研究[J].中国新药杂志,2010,19(12):1003-1006.
[2] Bergese S D, Puente E G. Clevidipine butyrate:A promising new drug for the management of acute hypertension[J].Expert Opin.Pharmacother,2010,11(2):281-295.
[3] Jung J S, You H H, Myn B. Synthesis of 1,4-dihydropyridine carboxylic acid(Ⅲ)[J].Arch Pharm Rse,1991,14(4):359-363.
[4] 宋志勇,沈国兵,刘艳华,等. 丁酸氯维地平中四种杂质的合成[J].中国药物化学杂志,2015,25(4):295-297.
[5] 林秀云,李银峰,陈晓雨,等. RP-HPLC法测定丁酸氯维地平原料药中有关物质[J].现代药物与临床,2015,30(12):1446-1450.
[6] Andersson K H, Nordlander M, Westerlund R C. Short-acting dihydropyridines:WO 9512578[P].1995.
[7] 高辉,钟静芬,时惠麟. 丁酸氯维地平合成路线图解[J].中国医药工业杂志,2009,40(10):791-793.
[8] 陈纪,张爱明,朱雪焱,等. 丁酸氯维地平的合成[J].中国新药杂志,2012,21(20):2368-2370.
[9] Mattson A, Svensson C, Thornblom K,etal. Manufacturing process:WO 0031035[P].2000.
Synthesis of The Degradation Impurities of Clevidipine Butyrate
CHU Xiao-qin1,2, YAN Shao-jie2, LI Shu-jun2, ZHANG Yuan2, HUANG Han-zhong2*, WANG Ping-bao2*
(1. Graduate School, Tianjin Medical University, Tianjin 300070, China; 2. Centre for Chemical Pharmaceutical Research, Tianjin Institute of Pharmaceutical Research Co., Ltd., Tianjin 300193, China)
Five degradation impurities of clevidipine butyrate, 4-(2,3-dichlorophenyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid monomethyl ester(A), 4-(2,3-dichlorophenyl)-1,4-dihydro-2,6-dimethy-3-pyridinecarboxylic acid methyl ester(B), 4-(2,3-dichlorophenyl)-2,6-dimethyl-3,5-pyridnedicarboxylic acid monomethyl ester(C), 4-(2,3-dichlorophenyl)-2,6-dimethyl-3,5-pyridinedicarboxylic acid methyl(1-oxobutoxy) methyl ester(D) and 4-(2,3-dichlorophenyl)-2,6-dimethyl-3-pyridinecarboxylic acid methyl ester(E), were synthesized using 4-(2,3-dichlorophenyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid (2-cyano-ethyl) methyl ester(5) as the starting material. A was synthesized by hydrolysis of 5, B was prepared by decarboxylation of A, C was synthesized by the hydrolysis after oxidation of 5, D was the condensation product of C and chloromethyl butyrate, and E was directly prepared by decarboxylation of C. The structures were confirmed by1H NMR and MS(ESI).
clevidipine butyrate; degradation impurity; drug synthesis
2016-04-12;
2016-11-08
褚小琴(1990-),女,汉族,湖北孝感人,硕士研究生,主要从事药物合成的研究。 Tel. 022-23003212, E-mail: chuxiaoqin2013@sina.com
黄汉忠,研究员, E-mail: huanghz@tjipr.com; 王平保,研究员,博士生导师, E-mail: wangpb@tjipr.com
R914.5; O621.3
A
10.15952/j.cnki.cjsc.1005-1511.2017.01.16105
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:24
上海计量测试(2020年1期)2020-03-18 02:31:22
中国资源综合利用(2017年4期)2018-01-22 02:46:55
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
化工进展(2015年6期)2015-11-13 00:27:25
环境科技(2015年1期)2015-11-08 12:10:50
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
无机化学学报(2014年8期)2014-02-28 17:32:44
无机化学学报(2014年7期)2014-02-28 17:32:21
中国氯碱(2014年10期)2014-02-28 01:04:59
