
V2O5/γ-Al2O3表面酸性和钒价态的调控及其异丁烷脱氢性能
2017-01-21 02:19徐华龙
石油化工 2016年9期
张 林,秦 枫,黄 镇,沈 伟,徐华龙
(复旦大学 化学系 上海市分子催化与功能材料重点实验室 复旦大学先进材料实验室,上海 200433)
V2O5/γ-Al2O3表面酸性和钒价态的调控
及其异丁烷脱氢性能
张 林,秦 枫,黄 镇,沈 伟,徐华龙
(复旦大学 化学系 上海市分子催化与功能材料重点实验室 复旦大学先进材料实验室,上海 200433)
以γ-Al2O3为载体,采用等体积浸渍法制备了负载高分散氧化钒催化剂(V2O5/γ-Al2O3),并通过K,Ca,Mg对V2O5/ γ-Al2O3的表面酸性及钒价态进行调控,研究催化剂表面酸性改性的变化规律及与异丁烷脱氢活性和选择性之间的构效关系。采用XRD、XPS、N2吸附-脱附、NH3-TPD和H2-TPR等方法对催化剂进行表征。实验结果表明,当氧化钒负载量低于7%(w),氧化钒在氧化铝表面处于良好分散状态时,催化剂保持最佳的反应活性和选择性。表征结果显示,碱金属K对V2O5/ γ-Al2O3表面的酸性和V的还原价态具有显著的调节作用,当K添加量(w)为1.5%时,强酸中心几乎被全部抑制,同时保留了适量的弱酸中心,而V的还原价态由2.87+调至3.95+;在1.5%K-V2O5/γ-Al2O3催化剂上异丁烷脱氢反应340 min后,异丁烷转化率保持在30%以上,异丁烯选择性保持在97%以上。
V2O5/γ-Al2O3催化剂;表面酸性;钒价态;异丁烷脱氢
低碳烷烃催化脱氢合成低碳烯烃是化石能源高值转化的重要路径之一,高效和稳定催化剂的研发是该工艺路线成功的关键点。目前,异丁烷脱氢主要采用Pt-Sn或Cr2O3/Al2O3催化剂,载体的表面酸中心位对催化活性起重要作用,反应温度通常在555~649 ℃[1-4],在该反应温度下脱氢产物异丁烯非常容易在强酸性位发生异构化、裂解、芳构化和聚合等副反应,导致催化剂的活性和选择性下降[5]。为了抑制这类现象发生,工业中采取了添加碱金属来调变催化剂表面酸性的措施,如在FBD-4工艺中,在铬催化剂中添加约2%(w)的K;在Oleflex工艺中,将Cs掺杂在Pt-Sn催化剂中[4]。但由于Cr和Pt基催化剂存在环境和经济性问题,在工业化应用中将会受到很大制约。
近年来,钒基催化剂应用于低碳烷烃脱氢和氧化脱氢合成烯烃的研究受到广泛关注。氧化脱氢具有转化率高和反应温度低的特点,但氧化反应深度不易控制,导致异丁烯的选择性较低[5]。Ogonowski等[6-7]发现在V2O5/Al2O3催化剂上,用二氧化碳作为温和氧化剂时,仍会导致异丁烯选择性的下降。相比较而言,低碳烷烃直接脱氢具有更好的选择性。Volpe等[8]通过对比了γ-Al2O3,α-Al2O3和USY沸石等载体,发现载体的酸性对脱氢的转化率和选择性具有显著影响。Harlin等[9]研究结果发现,酸性载体上钒催化剂的反应活性与氧化钒在表面的分散状态和价态有关,V3+和V4+均是异丁烷脱氢的活性中心。而Wang等[10]通过TPR和ESR等手段证明,V4+能稳定存在于VOx/Al2O3催化剂的表面。Fu等[11]发现,SnO2掺杂能提高氧化钒在氧化铝表面的分散状态,同时与钒物种的相互作用提高了催化剂的还原性,实现了催化性能的改善。由此可见,负载型钒基催化剂的氧化钒分散状态、表面酸性和钒价态直接关系到催化剂的活性和稳定性。
本工作以γ-Al2O3为载体,采用等体积浸渍法制备了负载高分散氧化钒催化剂(V2O5/γ-Al2O3),并通过K,Ca,Mg对V2O5/γ-Al2O3的表面酸性及钒价态进行调控,研究催化剂表面酸性改性的变化规律及与异丁烷脱氢活性和选择性之间的构效关系。采用XRD、XPS、N2吸附-脱附、NH3-TPD和H2-TPR等方法对催化剂进行表征。
1 实验部分
1.1 试剂及仪器
偏钒酸铵:AR,阿拉丁化学试剂上海有限公司;草酸:AR,上海强顺化学试剂有限公司;硝酸钾:AR,国药集团化学试剂有限公司;γ-Al2O3载体:国药集团化学试剂有限公司。
采用Bruker公司AXS D8 Advance型X射线衍射仪进行催化剂的XRD分析,CuKα射线,管电压40 kV,管电流40 mA,PSD检测;采用Perkin-Elmer公司PHI 5000 ESCASystem型X射线光电子能谱仪进行催化剂的XPS分析,AlKα射线,通能为46.95 eV;采用Micromeritics公司Tri Star 3000型自动物理吸附仪进行催化剂的N2吸附-脱附测定,测量前将催化剂在真空、160 ℃下预处理2 h,再于液氮温度下,用高纯氮进行吸附和脱附测定;采用Micromeritics公司Auto Chem 2920型氨程序升温脱附装置进行NH3-TPD测试,试样在高纯He气氛、550 ℃下吹扫2 h,冷却至室温,通入过量NH3直至吸附饱和,继续吹扫2 h以除去物理吸附的NH3,脱附过程由室温以10 ℃/min的速率升至500 ℃;采用Micromeritics公司ChemSorb2720型化学吸附仪进行H2-TPR测定,将约0.1 g试样装入U形石英管内,He气氛、600 ℃下预处理1 h,冷却至20 ℃,切换至H2/Ar混合气后,以10 ℃/min的升温速率在20~980 ℃区间记录H2还原信号;采用Thermo Fisher公司Thermo Trace Ultra GC型气相色谱仪对产物气体实现在线全分析,经气动六通阀自动采样,FID检测,Varian Plot Q色谱柱(50 m×0.32 mm×10 µm)。
1.2 催化剂的制备
称取一定量的偏钒酸铵和草酸于烧杯中,偏钒酸铵和草酸的摩尔比为1:2。加入一定量的蒸馏水(其量约为γ-Al2O3饱和吸附量的1.2倍),在80 ℃下搅拌至草酸和偏钒酸铵全部溶解。在溶液中加入额定量的γ-Al2O3载体,在缓慢搅拌中实现均匀浸渍,然后在静置状态下浸渍12 h。浸渍后载体于120 ℃下烘干,再于550 ℃下焙烧4 h。以该方法制得的催化剂标记为a%VOx/γ-Al2O3,其中,a为3,5,7,9,11,分别表示钒占催化剂的质量分数。
以7%VOx/γ-Al2O3催化剂为母体,以硝酸钾为前体,采用等体积浸渍方法对γ-Al2O3负载氧化钒催化剂进行改性。称取一定量的硝酸钾,量取与母体催化剂等体积的去离子水在室温下进行溶解,然后将溶液缓慢滴加于7%VOx/γ-Al2O3催化剂中。浸渍钾后的催化剂于120 ℃下烘干,然后在空气气氛、550 ℃下焙烧4 h。以该方法制得的催化剂标记为b%K-7%VOx/γ-Al2O3,其中,b=0.5,1.0,1.5,2.0,分别表示K占催化剂的质量分数。采用同样方法制备的Ca和Mg改性催化剂分别标记为1.5%Ca-7%VOx/γ-Al2O3和1.5%Mg-7%VOx/γ-Al2O3。
1.3 催化剂性能的评价
将上述催化剂应用于异丁烷催化脱氢制备异丁烯反应,采用全自动连续流动固定床反应装置评价催化剂性能。反应在常压下进行,反应管内径为8 mm,催化剂装填量为2.0 g。先在5%(φ)H2和95%(φ)N2气氛下,以10 ℃/min的速率将反应炉温度升到550 ℃,然后N2和原料气i-C4H10以15:1摩尔比混合进入反应管,反应床层温度由反应系统自动控制。
2 结果与讨论
2.1 V2O5在γ-Al2O3上的分散及其催化性能
氧化钒的表面分散状态对于脱氢催化剂的活性和稳定性有着重要的影响[12]。刘坚等[13]发现,以VOx/SBA-15为丙烷氧化脱氢催化剂,低钒负载量(w)(x<0.1%)可带来更高的醛类选择性,而中等的钒负载量则带来更高的丙烷脱氢深度氧化活性,高含量的钒负载量会抑制反应的活性。氧化钒同载体之间的相互作用也会影响氧化钒在催化剂表面的分散状态,带来脱氢活性的变化。由于氧化钒的塔曼温度低于400 ℃,在焙烧时会在载体表面发生自扩散现象,由于载体表面性质不同,氧化钒会产生不同的分散特性。Bond等[12]发现,当钒的负载量处在一个合适的范围时,钒能够在载体上呈最大程度的单层分散,而超过了这一界限,钒物种在表面会发生堆积覆盖,进而形成氧化钒结晶,导致催化剂活性降低。Höj等[14]通过喷雾热解法研究发现,随着负载量的升高,氧化钒由分散分布过渡到低聚分散,最终在负载量超过10%后出现晶粒。王珏等[15]采用XRD方法对金属氧化物在HZSM-5表面的MgO单层分散阈值进行了研究。本工作应用类似的方法将不同负载量的氧化钒负载在氧化铝载体上,然后应用XRD对载体表面的氧化钒的分散状态进行了表征。
图1为负载量3%~9%的a%VOx/γ-Al2O3系列催化剂的XRD谱图。由图1可知,在负载量低于7%时,XRD图谱中只出现了γ-Al2O3的衍射信号,未观察到氧化钒物种的衍射峰,这表明氧化钒在载体表面处于良好的分散状态;当钒负载量到达9%时,氧化钒物种的衍射峰开始出现。这表明氧化钒物种已经在载体表面发生多层负载,导致氧化钒晶体的聚集。因此,当氧化钒负载量低于7%时,氧化钒在γ-Al2O3表面处于良好的分散状态。
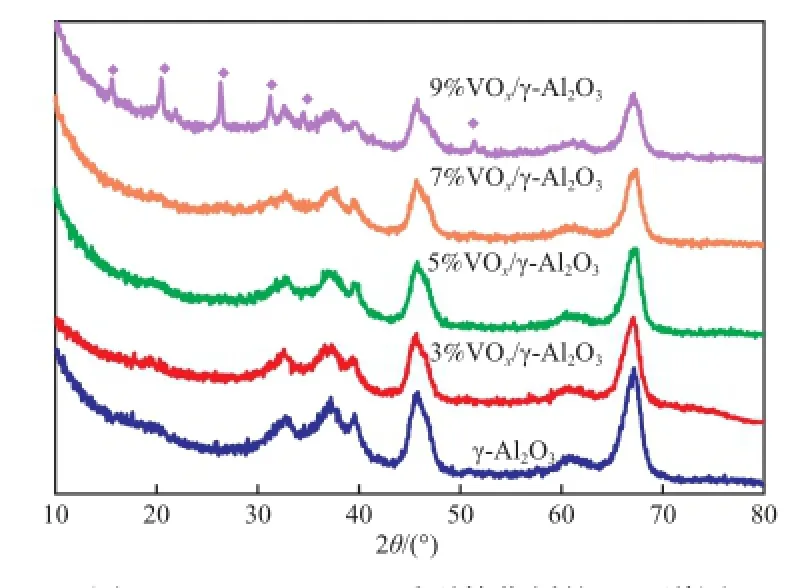
图1 a%VOx/γ-Al2O3系列催化剂的XRD谱图Fig.1 XRD patterns ofa%VOx/γ-Al2O3catalysts. a:the mass content of vandium in the catalyst.◆ V2O5
表1为a%V2O5/γ-Al2O3系列催化剂的比表面积和反应活性数据。由表1可见,随着钒负载量的升高,催化剂比表面积逐渐降低。当钒负载量在0~7%之间时,比表面积下降幅度较大,比表面积由210 m2/g降至170 m2/g,当钒负载量从7%增至9%时,比表面从170 m2/g略微下降至166 m2/g。该结果同样反映出氧化钒在γ-Al2O3表面的分散状态发生了显著改变。氧化钒在载体表面的分散状态直接关系到催化剂的反应性能,由表1给出的活性结果可见,氧化铝对于异丁烷脱氢没有活性,氧化钒负载量的变化对异丁烷脱氢的反应活性影响显著。7%VOx/γ-Al2O3催化剂具有最高的转化率和异丁烯选择性,分别为36.2%和88.1%。当氧化钒的负载量从3%提高至7%时,异丁烷的转化率逐渐升高,异丁烯选择性相对稳定。当氧化钒负载量为9%时,异丁烷的转化率降至23.1%,异丁烯选择性也出现明显的下降趋势。因此,异丁烷的催化脱氢活性与氧化钒在载体表面的分散状态密切相关,当氧化钒在氧化铝表面处于良好分散状态(7%VOx/γ-Al2O3)时,催化剂保持最佳的反应活性和选择性。

表1a%VOx/γ-Al2O3系列催化剂的比表面积和活性数据Table 1 Specifc surface area and activity of thea%V2O5/Al2O3catalysts
2.2 催化剂表征及其性能
2.2.1 K,Ca,Mg对催化剂表面酸性的调变作用
钒氧化物种在载体表面的分散状态和结构形式主要取决于载体的性质和负载量。Blasco等[16]研究了钒氧化物种在不同酸碱载体上的结构和分散状态,认为钒氧化物种在酸性载体表面易形成V2O5晶相,在碱性载体上易形成金属钒酸盐。Cortright等[17]研究发现,利用K2O对Pt-Sn/Al2O3催化剂改性可同时提高异丁烷的脱氢活性和选择性,其主要原因是通过碱金属元素改性一定程度上减少了酸性载体上活性金属的团聚,增加了催化剂活性表面。因此,钒氧化物种在载体表面的分散状态和表面酸性是决定异丁烷脱氢活性和稳定性的关键。本工作以γ-Al2O3负载高分散氧化钒为母体催化剂,通过等体积浸渍法对催化剂表面酸性进行调控,研究碱金属K和碱土金属Ca和Mg对催化剂表面酸性改性的变化规律。
采用NH3-TPD测定K对7%VOx/γ-Al2O3系列催化剂改性前后的NH3脱附温度和脱附量,结果见表2。由表2可知,在NH3脱附过程中,在160~171 ℃和630~645 ℃温区出现2个脱附峰,分别代表催化剂表面弱酸和强酸中心。可见,K的添加对催化剂表面酸量的抑制特别明显,当K添加量为1.0%时,强酸中心就几乎被全部中和,弱酸量仅为改性前的50%。经2.0%的K修饰后的7%VOx/γ-Al2O3表面总酸量从0.725 mmol/g下降至0.291 mmol/g。与此同时,NH3的脱附温度随着K添加量的增加向低温方向偏移,表明催化剂表面酸强度在减弱。由表2还可知,K改性使得催化剂的比表面积略有提高,但随着K添加量的提高比表面积呈下降趋势。当K添加量大于2.0%时,下降趋势显著增加。为了更好地研究负载型氧化钒表面酸性对反应性能的影响,分别制备了1.5%Ca和Mg改性的7%VOx/γ-Al2O3催化剂,并对催化剂表面酸性进行了对比研究。实验结果表明,相对于同样负载量的K改性,1.5%的Ca和1.5%的Mg改性的7%VOx/γ-Al2O3对催化剂表面酸性的调变作用较弱,改性后催化剂表面的强酸中心和弱酸中心有所下降,总酸量的下降幅度分别为22.8%和9.5%。NH3脱附峰温度变化不大,这表明碱金属和碱土金属元素都可实现对7%VOx/γ-Al2O3系列催化剂表面酸性的调变,对于催化剂表面酸量和酸强度的调节作用强弱次序为:K > Ca > Mg。

表2 催化剂的NH3-TPD表征结果Table 2 NH3-TPD results of some catalysts
2.2.2 K,Ca,Mg对催化剂中钒价态的调节作用
Wachs[18]通过拉曼、红外光谱等手段发现,K的加入能够改变V=O键的键合强度,这说明K元素不仅起到改变催化剂表面酸碱性的作用,在调节催化剂表面分散状态、电子性能等方面也起到一定作用。Harlin等[19]研究了钒氧化铝催化剂上V价态对于异丁烷脱氢的影响,研究结果表明:氧化钒在充分焙烧后主要以V5+存在,在异丁烷脱氢的过程中会被还原成更低的价态,催化剂的活性和V的还原价态有关,V的活性价态在3+和4+之间,在催化剂中添加Mg和Zr等金属能影响还原后V的价态,从而影响催化剂的转化率和选择性。
为了研究碱金属元素对V2O5/γ-Al2O3中V价态的调变作用,将不同添加量的K添加到7%VOx/ γ-Al2O3催化剂中,通过H2-TPR实验计算钒氧化态的变化。图2(A)是K改性前后7%VOx/γ-Al2O3催化剂的H2-TPR表征结果。由图2(A)可知,K的添加使得7%VOx/γ-Al2O3中氧化钒的还原程度显著降低,且随着K添加量的增加,还原峰面积逐步降低。从还原峰温度来看,催化剂的还原温度随K添加量的增加逐渐提高。结合表3中数据可知,当K的添加量为2.0%时,氧化钒的还原程度被严重抑制,其还原量仅为7%VOx/γ-Al2O3的50%。K改性导致催化剂还原量的降低说明催化剂表面V的氧化价态降低程度减小,氧化钒还原峰温的提高表明了氧化钒和载体之间的相互作用力得到增强。图2(B)比较了添加量为1.5%的K,Ca,Mg对7%VOx/ γ-Al2O3催化剂改性前后的H2-TPR表征结果。由图2(B)可知,同等添加量的K,Ca,Mg对7%VOx/ γ-Al2O3催化剂的改性均导致还原程度的降低,其中,Ca和Mg改性的催化剂H2还原量约为0.8 mmol/ g,略大于K改性的催化剂H2还原量(0.7 mmol/g),还原峰温的高低次序为:K>Ca>Mg。可见,碱金属K对7%VOx/γ-Al2O3催化剂的改性作用强于Ca和Mg。
根据Harlin等[19]报道的方法,将H2-TPR实验中的H2消耗量和V的价态进行关联,假设每摩尔的V5+价态降低至V4+价态将消耗0.5 mol的H2来计算还原后V的价态。表3为经不同K添加量改性后7%VOx/γ-Al2O3催化剂的H2还原量和还原后V的平均价态。由表3可知,未经改性的7%VOx/γ-Al2O3催化剂还原后,其V的价态为2.87+;经K改性后V的还原价态明显增加,当K的添加量在0.5%~1.5%之间时,V的平均价态在2.96+~3.95+价之间。同时还原峰温向高温方向移动,这表明了氧化钒和载体结合力增强。当K的添加量为2.0%时,V的平均价态为4.64+,还原峰温达到最大值545 ℃,表明当K的添加量较高时,也可能发生K与氧化钒在载体表面形成钒酸盐[16],导致了氧化钒还原程度的显著降低。由表3还可知添加量为1.5%的Ca和Mg改性的7%V2O5/γ-Al2O3催化剂还原后V的价态。添加量为1.5%的K改性后V的还原价态为3.95+,同样添加量的Ca和Mg改性后的V的还原价态为3.82+和3.83+。与K相比,Ca和Mg对V还原后价态的调节作用相对较弱。

图2 催化剂的H2-TPR表征结果Fig.2 H2-TPR results of the catalysts.a 7%VOx/γ-Al2O3;b 0.5%K-7%VOx/γ-Al2O3;c 1.0%K-7%VOx/γ-Al2O3;d 1.5%K-7%VOx/γ-Al2O3;e 2.0%K-7%VOx/γ-Al2O3;f 1.5%Ca-7% VOx/γ-Al2O3;g 1.5%Mg-7%VOx/γ-Al2O3
综上,碱金属和碱土金属对7%VOx/γ-Al2O3催化剂的改性作用不仅是对表面酸性的调控,还增强了氧化钒与载体的相互作用,提高了还原态V的价态,使V的价态处于具有更好催化性能的3+和4+之间。

表3 TPR法计算催化剂中V的平均氧化态Table 3 Average oxidation state of vanadium in the catalysts calculated by TPR
2.3 催化剂活性和稳定性
催化剂的异丁烷脱氢反应活性见表4。表4显示出K,Ca,Mg改性前后7%VOx/γ-Al2O3催化剂对异丁烷脱氢的活性评价结果。由表4可知,K改性对于催化剂的反应初活性有一定程度的抑制作用,当K的添加量为0.5%~1.5%时,异丁烷的转化率下降了约10百分点,但催化剂对于异丁烷转化的选择性显著提高,异丁烯选择性从原来的88%上升到97%左右。值得关注的是经190 min反应后,K-7%VOx/γ-Al2O3催化剂活性仍然保持稳定,异丁烷的转化率保持上升的趋势,高于异丁烷在7%VOx/γ-Al2O3催化剂上的转化率。随着反应的进行,异丁烯的选择性表现出不同的特征,在0.5%~1.0%K改性的7%VOx/γ-Al2O3催化剂上,经190 min反应后异丁烯的选择性有所下降,而经1.5%K改性的7%VOx/γ-Al2O3催化剂,异丁烯的选择性仍然保持在97%以上。但当K的添加量为2.0%时,转化率从改性前的36.2%下降到14.9%。造成这一现象的原因和催化剂表面酸中心的减少或是钒酸盐的存在密切相关。在同样反应条件下比较了1.5%Ca-7%VOx/γ-Al2O3和1.5%Mg-7%VOx/γ-Al2O3催化剂对异丁烷脱氢性能。研究结果表明,与K相比,负载量相同时Ca和Mg的改性作用略弱,反应初活性影响较小,选择性的增幅较小。负载量为1.5% 时,Ca和Mg改性催化剂异丁烷10 min转化率分别为30.1%和34.9%,高于K改性催化剂。经190 min反应后转化率分别保持在30.7%和28.7%。负载量为1.5% 时,Ca和Mg改性对催化剂的异丁烯选择性具有显著的提升作用,都保持在95%以上但略低于K改性催化剂的97.9%选择性。负载量相同时,Ca和Mg对7%VOx/γ-Al2O3催化剂的改性作用比K弱,这与催化剂的表征结果相一致。

表4 催化剂的异丁烷脱氢反应活性Table 4 Activities of the catalysts for the dehydrogenation of isobutane
图3为1.5%K-7%VOx/γ-Al2O3催化剂活性稳定性的评价结果。

图3 催化剂稳定性的评价结果Fig.3 Stability of the catalysts.● Selectivity to isobutene on 1.5%K-7%VOx/γ-Al2O3;■ Selectivity to isobutene on 7%VOx/γ-Al2O3;▼ Conversion of isobutane on 1.5%K-7%VOx/γ-Al2O3;▲ Conversion of isobutane on 7%VOx/γ-Al2O3
由图3可知,经负载量为1.5%的K改性的7%VOx/γ-Al2O3催化剂虽然初活性比7%VOx/ γ-Al2O3催化剂活性低,但在340 min反应范围内保持了良好的稳定性能,反应的转化率保持在30%以上。而7%VOx/γ-Al2O3催化剂的活性持续下降,在反应110 min后其转化率已经低于1.5%K-7% VOx/ γ-Al2O3催化剂,经340 min后反应转化率下降至16%。1.5%K-7%VOx/γ-Al2O3催化剂对于异丁烯的选择性普遍高于改性前催化剂,异丁烯的选择性保持在97%以上。
K,Ca,Mg改性的V2O5/γ-Al2O3催化剂的活性与催化剂表面酸性和V价态密切相关。马红超等[4]采用ESR技术对负载性V2O5/γ-Al2O3催化剂焙烧和还原后V的超精细结构进行了研究。研究结果表明,V4+超精细结构与活性物种在γ-Al2O3表面的分散有对应关系。Harlin等[6]应用XPS技术对V2O5/γ-Al2O3的还原价态进行了深入研究,指出焙烧后的氧化钒主要以V5+的形式存在,经H2还原后价态主要在V4+~V3+之间,异丁烷的脱氢活性与氧化钒的还原价态有密切关系。
综上, K对V2O5/γ-Al2O3催化剂表面的酸量和酸强度有显著的调节作用,极大降低了催化剂表面的强酸性位,这可有效抑制副反应和积碳,使催化剂具有更好的选择性和稳定性,但抑制副反应的同时也会使催化剂的初始转化率降低。H2-TPR表征结果显示,当K的添加量为1.5%时,催化剂表面的强酸位基本被中和,但仍具有相当量的弱酸位,同时V的还原价态为3.95+,这使得催化剂具有最佳的反应活性和选择性。Ca和Mg同样可对催化剂的表面酸性和钒还原价态进行调控,但与K相比,其调控作用略弱。
3 结论
1) 异丁烷的催化脱氢活性与氧化钒在载体表面的分散状态密切相关,当氧化钒负载量低于7%且氧化钒在氧化铝表面处于良好分散状态时,催化剂保持最佳的反应活性和选择性。
2) K对V2O5/γ-Al2O3催化剂表面的酸性和V的还原价态具有显著的调节作用,表面强酸位的减少和V还原价态的提高,可有效地提高催化剂的选择性和稳定性;当K添加量为1.5%时,强酸中心几乎被全部抑制,同时保留了适量的弱酸中心,而V的还原价态由2.87+调变为3.95+,这使催化剂的性能得到显著提高;在1.5%K-V2O5/γ-Al2O3催化剂上异丁烷脱氢反应340 min后,异丁烷转化率保持在30%以上,异丁烯选择性保持在97%以上。
3) Ca和Mg同样可以对V2O5/γ-Al2O3催化剂的表面酸性和钒还原价态进行调控,但与K相比,其调控作用略弱。
[1]Cortright R D,Dumesic J A. Microcalorimetric,spectroscopy,and kinetic-studies of silica-supported PT and PT/SN catalysts for isobutane dehydrogenation[J]. J Catal,1994,148(2):771 - 778.
[2]Cortright R D,Levin P E,Dumesic J A. Kinetic studies of isobutane dehydrogenation and isobutene hydrogenation over Pt/Sn-based catalysts[J]. Ind Eng Chem Res,1998,27(5):1717 - 1723.
[3]Bhasin M,McCain J,Vora B,et al. Dehydrogention and oxydehydrogenation of parrifns to olefns[J]. Appl Catal,A, 2001,221(1):397 - 419.
[4]Nawaz Z. Light alkane dehydrogenation to light olefn technologies:A comprehensive review[J]. Rev Chem Eng,2015,31(5):413 - 436.
[5]Carrero C A,Schloegl R,Wachs I E,et al. Critical literature review of the kinetics for the oxidative dehydrogenation of propane over well-defned supported vanadium oxide catalysts[J]. ACS Catal,2014,4(10):3357 - 3380.
[6]Ogonowski J,Skrzynska E. Dehydrogenationof isobutane in the presence of carbon dioxide over supported vanadium oxide catalysts[J]. React Kinet Catal Lett,2006,88(2):293 - 300.
[7]Vora B V. Development of dehydrogenation catalysis and processes[J]. Top Catal,2012,55(19):1297 - 1308.
[8]Volpe M,Tonetto G,de Lasa H. Butane dehydrogenation on vanadium supported catalysts under oxygen free atmosphere[J]. Appl Catal,A,2004,272(1/2):69 - 78.
[9]Harlin M E,Niemi V M,Krause A O I. Alumina-supported vanadium oxide in the dehydrogenation of butanes[J]. J Catal,2000,195(1):67 - 68.
[10]Wang Guojia,Ma Hongchao,Li Ying,et al. Dehydrogenation of isobutane over V2O5/gamma-Al2O3catalyst[J]. React Kinet Catal Lett,2001,74(1):103 - 110.
[11]Fu Yinghuan,Ma Hongchao,Wang Zhenlu,et al. Charaterization and reactivity of SnO2-doped V2O5/gamma-Al2O3catalysts in dehydrogenation of isobutane to isobutene[J]. J Mol Catal,A:Chem,2004,221(1/2):163 - 168.
[12]Bond G C,Tahir S F. Vanadium-oxide monolyer catalysts-preparation,characterization and catalytic activity[J]. Appl Catal,1991,71(1):1 - 31.
[13]刘坚,赵震,王宏宣,等. 担载钒氧化物催化剂对丙烷选择氧化性能[J]. 物理化学学报,2011,27(11):2659 - 2664.
[14]Höj M,Jensen A D,Grunwaldt J. Structure of alumina supported vanadium catalysts for oxidative dehydrogenation of propane prepared by fame spray pyrolysis[J]. Appl Catal,A,2013,451,207 - 215.
[15]王珏,赵碧英,谢有畅. MgO/HZSM-5中MgO分散状态和催化性能的关系[J]. 物理化学学报,2001,17(11),966 -971.
[16]Blasco T,Lopez N J M. Oxidative dehydrogenation of short chain alkane on supported vanadium oxide catalysts[J]. Appl Catal,A,1997,157(1/2):117 - 142.
[17]Cortright R D,Dumesic J A. Efect of potassium on silica-supported Pt and Pt/Sn catalysts for isobutane dehydrogenation[J]. J Catal,1995,157(2):576 - 583.
[18]Wachs I E. Recent conceptual advances in the catalysis science of mixed oxide catalytic materials[J]. Catal Today,2005,100(1/2):79 - 94.
[19]Harlin M,Niemi V,Krause A,et al. Effect of Mg and Zr modifcation on the activity of VOx/Al2O3catalysts in the dehydrogenation of butanes[J]. J Catal,2001,203(1):242 -252.
(编辑 杨天予)
Modification of acidity and vanadium valence of V2O5/γ-Al2O3catalyst for dehydrogenation of isobutane
Zhang Lin,Qin Feng,Huang Zhen,Shen Wei,Xu Hualong
(The Department of Chemistry,Fudan University,Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials,Laboratory of Advanced Materials of Fudan University,Shanghai 200433,China)
Highly dispersed V2O5/γ-Al2O3catalysts for the dehydrogenation of isobutane were prepared through impregnation,and K,Ca and Mg were introduced to modify the surface acidity and vanadium valence of the V2O5/γ-Al2O3catalysts. The catalysts were characterized by means of XRD,XPS,N2adsorption and desorption, NH3-TPD,and H2-TPR. It was indicated that,when the vanadium loading was less than 7%(w),the vanadium oxide dispersed well on the γ-Al2O3support and the catalysts showed high activity in the dehydrogenation of isobutane to isobutene. The characterization results revealed that the potassium modification had significant adjustment ef ects on the surface acidity and vanadium valence of the catalysts. The strong acid sites were nearly neutralized when the loading of potassium was 1.5%(w),while the moderate amount of weak acid sites was kept on the catalyst surface and the vanadium valence was adjusted from 2.87+ to 3.95+. After the dehydrogenation over the 1.5%(w)K-V2O5/γ-Al2O3catalyst was conducted for 340 minutes,the conversion of isobutane and the selectivity to isobutene were still kept above 30% and more than 97%,respectively.
V2O5/γ-Al2O3catalyst;surface acidity;vanadium valence;isobutane dehydrogenation
1000 - 8144(2016)09 - 1043 - 07
TQ 223.12
A
10.3969/j.issn.1000-8144.2016.09.004
2016 - 03 - 17;[修改稿日期]2016 - 06 - 15。
张林(1988—),男,山东省烟台市人,硕士生,电话 021 - 65642401,电邮 11210220070@fudan.edu.cn。联系人:徐华龙,电话 021 - 65642401,电邮 shuhl@fudan.edu.cn。
上海市科学技术委员会资助国际合作项目(14120700700);重点实验室基金项目(14DZ2273900)。
猜你喜欢
化学工程师(2023年1期)2023-02-17
上海金属(2022年5期)2022-09-26
理化检验-化学分册(2020年12期)2021-01-26
福建基础教育研究(2019年8期)2019-05-28
上海农业科技(2019年1期)2019-02-22
中国测试(2018年4期)2018-05-14
中国果业信息(2018年5期)2018-01-17
中学生数理化·八年级物理人教版(2017年6期)2017-11-09
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
医学研究杂志(2015年3期)2015-06-10
