
TGF-β Ⅲ型受体介导的信号通路及其在纤维化疾病中的作用
2017-01-06 08:09厉歆然孙妩弋谷元婧彭文婷
中国药理学通报 2016年12期
厉歆然,孙妩弋,谷元婧,彭文婷,魏 伟
(安徽医科大学临床药理研究所,抗炎免疫药物教育部重点实验室,抗炎免疫药物安徽省协同创新中心, 安徽 合肥 230032)
◇讲座与综述◇
TGF-β Ⅲ型受体介导的信号通路及其在纤维化疾病中的作用
厉歆然,孙妩弋,谷元婧,彭文婷,魏 伟
(安徽医科大学临床药理研究所,抗炎免疫药物教育部重点实验室,抗炎免疫药物安徽省协同创新中心, 安徽 合肥 230032)
转化生长因子-β(transforming growth factor-β, TGF-β)在细胞的自我调控包括细胞增殖、分化、凋亡、胚胎发育、免疫监督、血管再生等过程中发挥重要作用。TGF-βⅢ型受体(TβRⅢ)是 TGF-β超家族的辅助受体之一,TβRⅢ不仅能介导TGF-β超家族配体依赖的Smad经典通路,还可以介导非Smad依赖的信号通路,参与调控细胞的多种信号通路,在纤维化、肿瘤、心血管系统等疾病中发挥重要作用。该文就TβRⅢ参与的信号通路及其在纤维化疾病中的作用作一综述。
TGF-βⅢ型受体;纤维化疾病;信号通路;β-arrestin2;Smad;MAPK
转化生长因子(transforming growth factor-β, TGF-β)是一类结构相似、功能相近的多肽组成的细胞因子,属于调节细胞生长和分化的TGF-β超家族分子。在哺乳动物体内,TGF-β超家族参与调节多种细胞功能,包括细胞生长、分化、凋亡、胶原生成、免疫反应以及胚胎的生长[1]。TGF-β包含3种高度相关的细胞表面受体——TGF-βⅠ型受体(type Ⅰ TGF-β receptor,TβRⅠ)、TGF-βⅡ型受体(TβRⅡ)、TGF-βⅢ型受体(TβRⅢ)[2]。TβRⅢ是TGF-β超家族的辅助受体,既可以介导经典的Smad信号通路,又可以介导非经典Smad信号通路。近年的相关研究表明,TβRⅢ通过其参与调节的各种信号通路,在纤维化、肿瘤、心血管系统等疾病中发挥重要作用。纤维化是由慢性组织损伤导致的细胞外基质(extracellular matrix, ECM)的过度沉积。纤维化疾病发病人群众多,患者往往有多个组织器官受累,尚缺乏有效的治疗方法。研究表明,TβRⅢ与纤维化疾病有着密切的关系,并在纤维化疾病中发挥着重要作用。本文就TβRⅢ介导的信号通路及其在纤维化疾病中作用的研究进展作一综述。
1 TβRⅢ的基因位点及蛋白结构
1.1 TβRⅢ的基因位点 TβRⅢ几乎表达于所有种类的细胞中,且是这些细胞中表达最丰富的TGF-β超家族受体,每个细胞至少表达2×105个TβRⅢ受体,而大多数Ⅰ型、Ⅱ型受体只表达5 000~10 000个。编码TβRⅢ的基因位于染色体1p31-32,包含16个外显子、2个启动子,其中1个近端启动子、1个远端启动子,这使得可以产生两种mRNA。在大多数组织类型中,主要利用近端启动子开启转录[3]。
1.2 TβRⅢ的蛋白结构
1.2.1 TβRⅢ基本结构 TβRⅢ以非共价连接的同源二聚体表达于细胞表面[3]。它由851个氨基酸构成的跨膜蛋白多糖组成,其中包括1个含766个氨基酸的胞外域,1个疏水跨膜域,以及1个缺乏酶活性含42个氨基酸的胞内域。TβRⅢ胞外域包含1个氨基端独立结构域、1个可能与受体的寡聚化反应有关的透明带结构域(zona pelucida domain,ZPD),还包括两个独立的TGF-β配体结合域,其中一个为靠近氨基端的远端结合域,另一个为靠近羧基端的近端结合域[3-4]。TβRⅢ通过配体结合域结合TGF-β1、TGF-β2、TGF-β3、骨形态发生蛋白(bone morphogenetic protein,BMP) -2、4、7、生长分化因子5(growth differentiation factor 5,GDF-5)和抑制素A,另外通过糖胺聚糖(glycosaminoglycan, GAG)侧链的结构修饰与成纤维细胞生长因子 (fibroblast growth factor,FGF)结合[5]。尽管这些结构域有重叠,比如抑制素结合域完全包含于TβRⅢ近膜区域的ZPD结构中[6],但是TβRⅢ拥有独立的抑制素和TGF-β结合域。TβRⅢ的结构如Fig1所示。
1.2.2 TβRⅢ基本结构的功能 TβRⅢ翻译后的结构修饰对TβRⅢ的功能很重要。TβRⅢ核心蛋白的分子质量被认为是100 ku,然而由于GAG侧链的翻译后修饰,完全加工后的TβRⅢ的分子质量则为180~300 ku[3]。在缺失内源性TβRⅢ的猪肾脏上皮细胞LLC-PK1中,表达GAG侧链高度修饰的TβRⅢ,可以阻止TβRⅠ和TβRⅡ形成复合物,抑制TGF-β信号的传导,从而抑制TGF-β1诱导的LLC-PK1细胞的生长和胶原沉积[7]。TβRⅢ的GAG侧链修饰可能对TβRⅢ调控细胞迁移的能力也有贡献,例如无论在卵巢癌细胞Ovca429还是在正常卵巢上皮细胞中,缺失GAG侧链的TβRⅢ突变体都会减弱TβRⅢ对细胞迁移的抑制[8]。
TβRⅢ含有一段短的缺乏固有酶活性的胞质域,此结构域中还包含一段PDZ一级结合域[9-10]。PDZ的名称来源于最早发现的含有此结构域的3个蛋白质:突触后密度蛋白(postsynaptic density protein, PSD-95)、肿瘤抑制蛋白(disc large tumor suppressor gene,DLG)、紧密连接蛋白(zona occludens-1,ZO-1)的首字母。PDZ一级结合域能与众多的蛋白相互作用,在信号转导、蛋白质的定位等方面发挥重要功能[11]。删除胞质域的TβRⅢ仍然可以结合TGF-β1配体,并呈递给TβRII,但它失去了呈递TGF-β2的能力[10]。尽管TβRⅢ的细胞质结构域对配体的呈递并非是至关重要的,但它被认为在调控TGF-β介导的信号通路和生长抑制方面发挥至关重要的作用。TβRⅢ胞质域841位的苏氨酸被TβRⅡ(其本身具有激酶活性)磷酸化,使其可与支架蛋白β-arrestin2相互作用。此位点的磷酸化是TβRⅢ与β-arrestin2结合所必需的,突变体TβRⅢT841A(841位的苏氨酸突变为丙氨酸)将彻底取消两者之间的相互作用。TβRⅢ的胞质域与β-arrestin2相互作用导致β-arrestin2/TβRⅡ/TβRⅢ复合物以囊泡形式内吞,并下调TGF-β信号[12]。
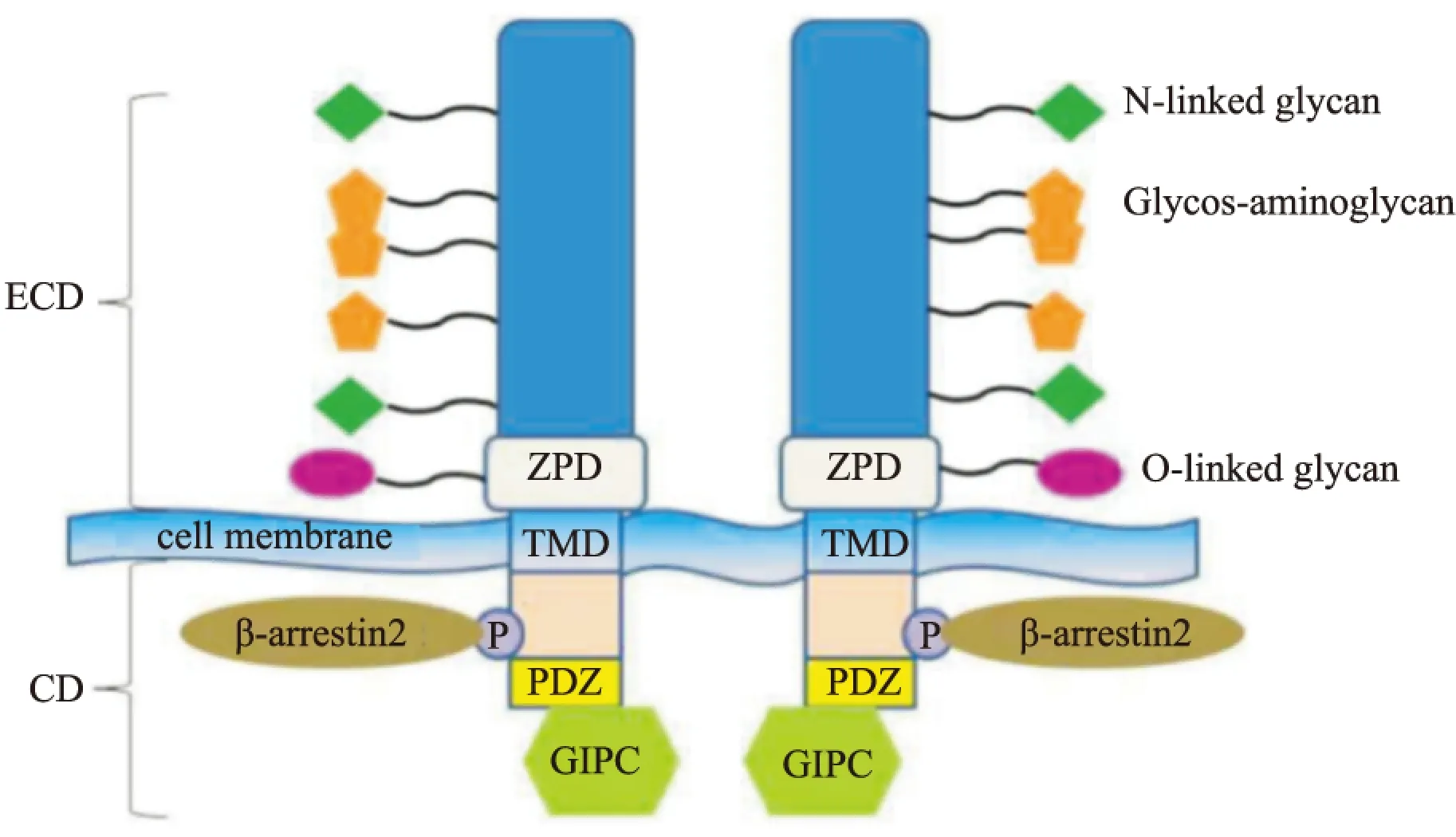
Fig 1 Structure of TβRⅢ
ECD: extracellular domain;CD: cytoplasmic domain;TMD: transmembrane domain;GIPC: G alpha interacting protein C terminus,Gα;PDZ: PSD-95、DLG、ZO-1
Gα相互作用蛋白C末端(G alpha interacting protein C terminus,GIPC)是1个含333个氨基酸的蛋白分子,TβRⅢ通过胞质域的PDZ一级结合域也可与GIPC相互作用。与GIPC相互作用可以使TβRⅢ在细胞表面稳定表达,并增强其对TGF-β配体的结合能力。TβRⅢ和GIPC的相互作用是TβRⅢ的稳定所必需的,而TβRⅢ-DEL(1个缺乏PDZ结合域的突变体)不能够结合GIPC,因而不能稳定TβRⅢ的水平[9]。此外, GIPC还可以介导TβRⅢ与TβRⅠ、TβRⅡ形成聚合物并内吞,抑制TGF-β信号传导[13]。在乳腺癌中,TβRⅢ的胞质域与GIPC相互作用是TβRⅢ抑制细胞迁移、侵袭及乳腺癌细胞在体内转移必不可少的[14]。
2 TβRⅢ介导的信号通路
2.1 TGF-β经典信号通路 TβRⅢ在TGF-β经典信号通路中除了发挥传统的结合呈递配体的作用外,在许多疾病状态下,TβRⅢ还能够与TβRI和TβRII形成复合物,内吞到胞内,阻断TGF-β信号通路。TβRⅢ在TGF-β经典信号通路中发挥的作用如Fig 2所示。
TβRⅢ为表达最丰富的TGF-β超家族受体,最显著的特点就是作为超家族的共受体呈递多种TGF-β超家族的配体,包括TGF-β1、2、3,抑制素,BMP-2、4、7,GDF-5[5, 10, 15]。TβRⅢ呈递配体的能力对于TGF-β超家族的一些配体尤其重要,比如TGF-β2和抑制素都不能独立地与相应的受体结合,而需要通过TβRⅢ的呈递 。文献报道,内皮细胞可以通过表达TβRⅢ增强对TGF-β2信号的敏感性,脑垂体促肾上腺皮质瘤细胞和卵巢细胞能够通过表达TβRⅢ增强对抑制素的敏感性。另外,由于TβRⅢ可以通过促进抑制素与激活素Ⅱ型受体、BMPⅡ型受体结合,竞争性拮抗了激活素、BMPs与这两种受体的结合,从而抑制了激活素和BMP信号通路[15-16]。
TβRⅢ除了发挥配体的辅助呈递作用外,近年来研究发现其还可与TβRⅠ和TβRⅡ形成复合物,抑制TGF-β介导的Smad信号通路。最近的研究表明,配体TGF-β1与TβRⅢ结合后,TβRⅠ和TβRⅡ可以分别结合到TβRⅢ的不同结构域形成复合物,而这种结合是由GIPC或者β-arrestin2等蛋白分子与TβRⅢ的胞质域相互作用介导的,形成的复合物在β-arrestin2的作用下内吞,抑制TGF-β1介导的Smad2/3信号通路[12-13]。如果缺乏TβRⅢ的表达,TGF-β1直接结合TβRⅡ,并与TβRⅠ形成复合物,从而激活TGF-β介导的Smad信号通路[13]。在本身不表达TβRⅢ的恶性乳腺癌细胞4T1中转染正常TβRⅢ腺病毒,能明显抑制TGF-β介导的Smad信号通路,而转染TβRⅢ-del突变体,则不能抑制Smad信号通路。再向稳定表达TβRⅢ的4T1细胞中转染shGIPC质粒,则取消了之前对Smad信号通路的抑制作用[14]。同样,在HEK293细胞(不表达TβRⅢ)中转染TβRⅢT841A,β-arrestin2将不能介导TβRⅢ与TβRⅠ、TβRⅡ形成的复合物内吞,从而取消对Smad信号通路的抑制[12]。TβRⅢ还可以通过抑制TGF-β介导的Smad信号通路,抑制新生小鼠心肌纤维母细胞由缺氧引起的凋亡,并且可以减少缺氧的心肌纤维母细胞中p-Samd2/3、TβRⅠ/TβRⅡ复合物和胶原的产生[17]。
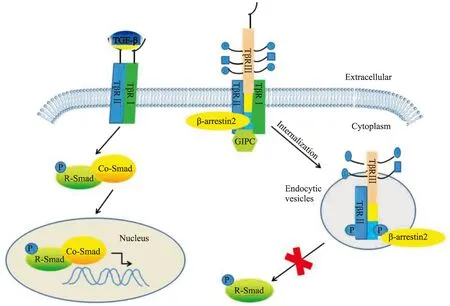
Fig 2 Role of TβRⅢ in classical TGF-β-Smad signaling pathway
2.2 TGF-β非经典信号通路 TβRⅢ除了可以介导TGF-β经典信号通路,参与体内许多生理或病理过程的改变,还可以在其他一些非经典通路中发挥作用。TβRⅢ介导的非经典信号通路如Fig 3所示。
2.2.1 NF-κB信号通路 在正常的乳腺上皮细胞和乳腺癌细胞中,TβRⅢ通过β-arrestin2负性调控NF-κB信号。过表达TβRⅢ可减少NF-κB介导的转录激活和IκB的降解,当用TβRⅢT841A突变体转染MCF10A和MDA-MB-231细胞时,由于TβRⅢ不能与β-arrestin2相互作用则没有这种调节能力[18]。在正常的乳腺上皮细胞中下调TβRⅢ的表达将导致NF-κB的活性增加,NF-κB介导的钙黏蛋白表达也随之下降,细胞的增殖、侵袭能力增强,这提示TβRⅢ通过调控钙黏蛋白的表达来保持上皮细胞的表型[18-19]。将TβRⅢ被下调的正常乳腺上皮细胞(NMuMG细胞)注射进裸鼠体内,10周后形成了具有侵袭能力的肿瘤,而注射正常乳腺上皮细胞的裸鼠则没有出现这种转变,表明在正常乳腺上皮细胞中TβRⅢ通过负性调节NF-κB信号降低细胞增殖、侵袭能力,抑制肿瘤的生成[19]。
2.2.2 MAPK信号通路 TβRⅢ被认为通过配体依赖的Smad3通路和配体非依赖的p38信号通路在调节生长抑制和凋亡方面发挥重要作用。在L6成肌细胞中过表达TβRⅢ,它既可以通过TGF-β1介导的Smad途径,又可以通过p38途径增强生长抑制作用。在肾透明细胞癌动物模型和体外培养细胞中,恢复TβRⅢ的表达可增加p38的磷酸化,通过非Smad依赖的p38 MAPK途径诱导肾透明细胞癌细胞的凋亡。TβRⅢ对p38的磷酸化作用至少部分依赖于TβRⅢ的胞外域[20]。另外,TβRⅢ还可以通过调节ERK信号通路发挥不同的生理作用。在小鼠肺纤维化细胞中TβRⅢ可以通过抑制ERK信号通路,减少α-SMA和Ⅰ型前胶原的表达,减轻肺纤维化[21]。而在内皮细胞中,TβRⅢ可以与β-arrestin2相互作用而抑制ERK信号通路,从而抑制内皮细胞的迁移[22]。
2.2.3 Akt信号通路 在小鼠肺纤维化细胞中表达外源性TβRⅢ可以抑制α-SMA和Ⅰ型前胶原的表达,以及Akt的磷酸化,提示TβRⅢ可以通过抑制Akt信号通路发挥减轻肺纤维化的作用[21]。TβRⅢ还可以通过抑制Akt信号通路调控肝癌细胞的侵袭、转移。在肝癌细胞恶化过程中,TβRⅢ的表达不断减少,而癌细胞的侵袭转移能力不断增强。相比于阴性对照组,转染了TβRⅢ siRNA的肝癌细胞SMMC-7721中p-Akt的表达明显增多,且细胞的侵袭转移能力明显增强[23]。 2.2.4 Cdc42信号通路 TβRⅢ可通过与β-arrestin2相互作用激活配体非依赖的Cdc42信号通路,调节细胞的迁移。 在卵巢表面上皮细胞中,缺乏配体诱导的条件下,下调TβRⅢ的表达可以改变肌动蛋白的细胞骨架,增加肌纤维和板状伪足的张力,从而增加细胞定向或随机的迁移。反之,增加TβRⅢ的表达可抑制正常上皮细胞和癌细胞的迁移,这主要通过持续地激活Cdc42信号通路,而不依赖经典的Smad信号通路[8]。TβRⅢ对细胞迁移的作用部分是由其GAG侧链和胞质域介导的,而TβRⅢ的突变体,比如缺乏GAG侧链的TβRⅢΔGAG和缺乏胞质域的TβRⅢΔcyto相比于完整的TβRⅢ则不能抑制迁移[8]。
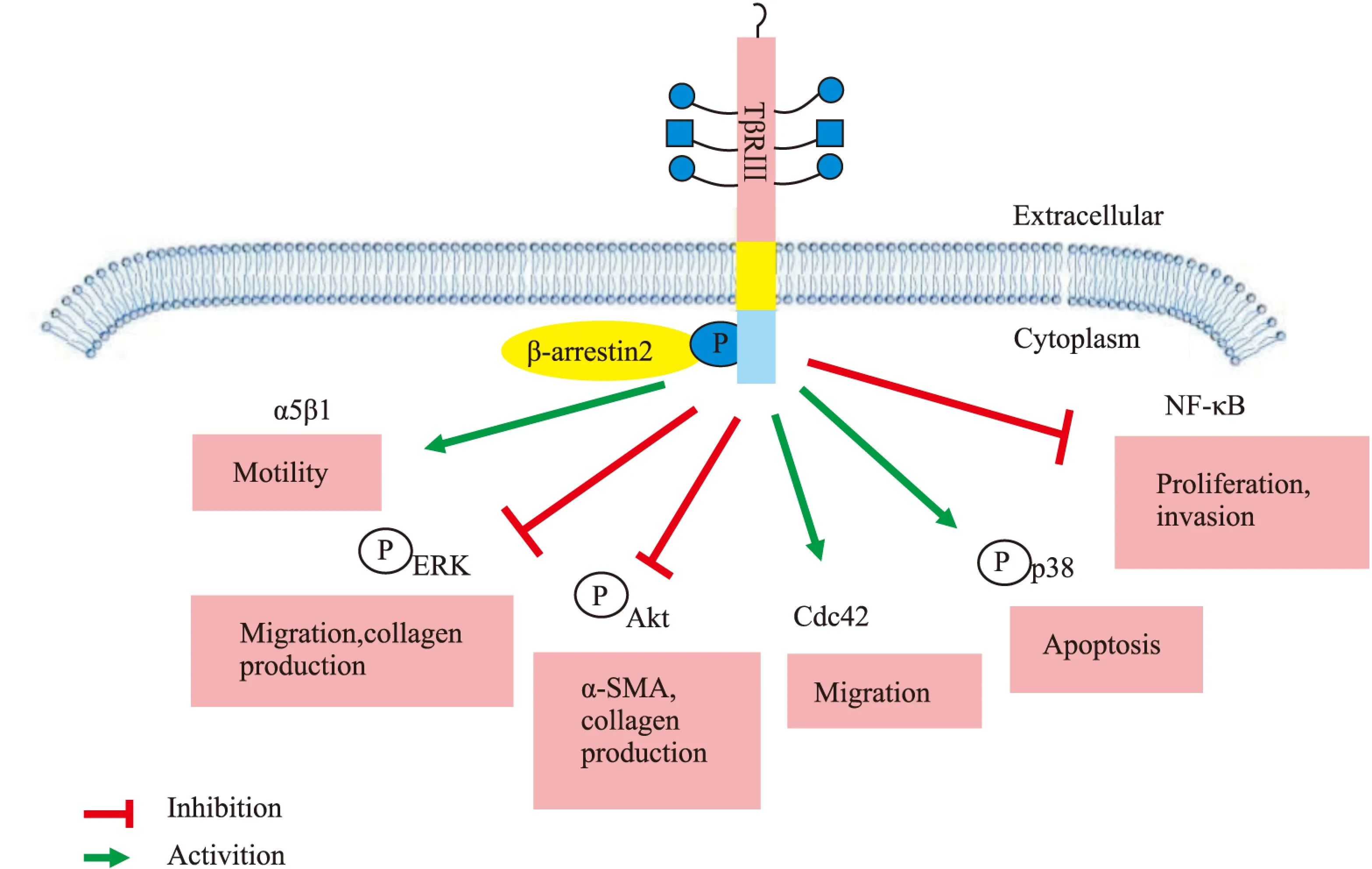
Fig 3 Role of TβRⅢ in non-Smad signaling pathways
2.2.5 整合素α5β1信号通路 在内皮细胞中,TβRⅢ与β-arrestin2相互作用可以介导活化的整合素α5β1的内吞迁移并与黏着斑结合,促进黏着斑的形成,以及使内皮细胞黏附纤维连接蛋白(fibronectin,FN)。进一步的研究发现,在乳腺癌组织中,TβRⅢ可以促进整合素α5在相互黏附的细胞之间定位,抑制肿瘤的恶性迁移,并且与肿瘤患者的整体生存率有关,可作为乳腺癌整体生存率的预测指标[24]。
3 TβRⅢ在纤维化疾病中的作用
纤维化是指由于炎症导致器官实质细胞发生坏死,组织内ECM异常增多和过度沉积的病理过程,可发生于多种器官,如肝脏、肾脏、胆囊等。目前,纤维化疾病的治疗仍然没有非常有效的方法,只能通过抑制 ECM 生成、加快 ECM 降解以及抵抗炎症作用来预防和减轻纤维化。近年来,越来越多的研究发现,TβRⅢ与纤维化的形成有着密切联系,影响着纤维化的进程。
3.1 心肌纤维化 心肌纤维化表现为心肌纤维母细胞异常增殖、ECM过度沉积。有研究发现[24],在由心肌梗死诱发的心肌纤维化小鼠心脏中TGF-β1的表达升高,而TβRⅢ的表达降低。向心肌纤维母细胞中转染TβRⅢ可以抑制胶原的生成,进一步的研究发现其还可通过减少TGF-β1和p-Smad3的表达,降低TGF-β信号通路的活性。另外,有研究发现[25],来源于TβRⅢ的合成肽P144在原发性高血压大鼠模型中也表现出抗心肌纤维化的特点,与模型组相比,在给予P144的实验组大鼠心肌细胞中p-Smad2、结缔组织生长因子(connective tissue growth factor,CTGF)、前胶原α1 (procollagen α1)和Ⅰ型胶原的mRNA和蛋白表达,以及胶原相关交联与沉积都明显降低。
3.2 肺纤维化 在肺纤维母细胞分化的过程中,给予TGF-β1刺激后TβRⅢ的表达被下调,而在小鼠肺纤维化细胞中表达外源性TβRⅢ可以抑制TGF-β1介导的纤维母细胞的分化,α-SMA和Ⅰ型前胶原的表达,以及Smad2/3、Akt、ERK的磷酸化[21]。可以看出,TβRⅢ能够抑制纤维母细胞的分化,且既通过TGF-β1介导的Smad 依赖途径,也通过非Smad依赖途径。另外有研究表明[26],肺纤维母细胞在TGF-β1的刺激下,给予糖皮质激素,可以增加TβRⅢ的表达,明显降低p-Smad2、p-Smad3的表达,但却能增加p-Smad1/5/8的表达,而转染TβRⅢ siRNA则明显减弱了这种作用。表明TβRⅢ具有辅助糖皮质激素调节TGF-β信号通路,减轻肺纤维化的作用。3.3 肝纤维化 体外培养肝星状细胞(hepatic stellate cells, HSC),发现在HSC转化为成纤维细胞的过程中,细胞内TβRⅢ的mRNA水平下降,TβRⅢ的表达减少[27]。在TGF-β1刺激的MV1Lu 细胞中加入来源于TβRⅢ的合成肽P144,能够明显抑制MV1Lu 细胞的增殖,同时阻碍TGF-β1与其受体的结合,而将P144腹腔注射于四氯化碳(CCl4)诱导的肝纤维化大鼠时,能够抑制HSC的激活,抑制Ⅰ型胶原的转录和α-SMA的表达,阻碍TGF-β1与其受体的结合,表现出有效的抗纤维化作用[28]。
3.4 肾纤维化 有研究表明[29],腹腔给予P144能够明显减少原发性高血压大鼠模型中肾纤维化过程中的各型胶原及结缔组织生长因子(connective tissue growth factor,CTGF)mRNA水平的增高,减轻肾纤维化症状。另外,细胞表面硫酸肝素蛋白多糖syndecan-2的核心蛋白S2ΔS可以与TGF-β1结合,封闭TGF-β信号通路,降低TβRI、TβRII的表达,增加TβRⅢ的表达,且免疫共沉淀的结果显示S2ΔS只与TβRⅢ有免疫共沉淀反应,而与Ⅰ、Ⅱ型受体无此反应[30]。此结果可能提示了S2ΔS在肾纤维化中通过增加TβRⅢ的表达而抑制TGF-β信号通路,减慢肾纤维化进程。
4 结语
曾经仅仅被认为是TGF-β辅助受体的TβRⅢ,在近些年的研究中,显现出越来越重要的地位,它不仅能介导TGF-β经典Smad信号通路,还能参与调控许多非Smad依赖的信号通路,在纤维化疾病中发挥着重要的作用。这些研究为寻找纤维化治疗的新靶点以及评估纤维化程度奠定了理论基础并指明了新的方向。然而,仍有许多问题还有待解决,比如TβRⅢ参与的这些信号通路与相关蛋白因子之间是怎样相互作用的?TβRⅢ还可以通过哪些分子机制调控机体的生理或病理改变?因此,对TβRⅢ参与调控信号通路的分子机制,以及如何开发靶向治疗药物都将成为我们今后不断努力探究的方向。
[1] 张 森, 孙妩弋, 吴晶晶, 等. TGF-β信号通路及以其为靶点的肝病治疗药物研究进展[J]. 中国药理学通报,2013,29(11):1489-93.
[1] Zhang S, Sun W Y, Wu J J, et al. Targeting the TGF-β signaling pathway in liver diseases[J].ChinPharmacolBull, 2013, 29(11):1489-93.
[2] Gatza C E, Oh S Y, Blobe G C. Roles for the type III TGF-beta receptor in human cancer[J].CellSignal, 2010, 22(8):1163-74.
[3] Lopez-Casillas F, Cheifetz S, Doody J, et al. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system[J].Cell, 1991, 67(4):785-95.
[4] Mendoza V, Vilchis-Landeros M M, Mendoza-Hernandez G, et al. Betaglycan has two independent domains required for high affinity TGF-beta binding: proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor[J].Biochemistry, 2009, 48(49):11755-65.
[5] Bernabeu C, Lopez-Novoa J M, Quintanilla M. The emerging role of TGF-beta superfamily coreceptors in cancer[J].BiochimBiophysActa, 2009, 1792(10):954-73.
[6] Wiater E, Harrison C A, Lewis K A, et al. Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan: functional separation of betaglycan co-receptor actions[J].JBiolChem, 2006, 281(25):17011-22.
[7] Eickelberg O, Centrella M, Reiss M, et al. Betaglycan inhibits TGF-beta signaling by preventing type Ⅰ-type Ⅱ receptor complex formation. Glycosaminoglycan modifications alter betaglycan function[J].JBiolChem, 2002, 277(1):823-9.
[8] Mythreye K, Blobe G C. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42[J].ProcNatlAcadSciUSA, 2009, 106(20):8221-6.
[9] Blobe G C, Liu X, Fang S J, et al. A novel mechanism for regulating transforming growth factor beta (TGF-beta) signaling. Functional modulation of type III TGF-beta receptor expression through interaction with the PDZ domain protein, GIPC[J].JBiolChem, 2001, 276(43):39608-17.
[10] Blobe G C, Schiemann W P, Pepin M C, et al. Functional roles for the cytoplasmic domain of the type III transforming growth factor beta receptor in regulating transforming growth factor beta signaling[J].JBiolChem, 2001, 276(27):24627-37.
[11] Dunn H A, Ferguson S S. PDZ protein regulation of G protein-coupled receptor trafficking and signaling pathways[J].MolPharmacol, 2015, 88(4):624-39.
[12] Chen W, Kirkbride K C, How T, et al. Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signaling[J].Science, 2003, 301(5638):1394-7.
[13] Tazat K, Hector-Greene M, Blobe G C, Henis Y I. T betaRⅢ independently binds type I and type II TGF-beta receptors to inhibit TGF-beta signaling[J].MolBiolCell, 2015, 26(19):3535-45.
[14] Lee J D, Hempel N, Lee N Y, Blobe G C. The type III TGF-beta receptor suppresses breast cancer progression through GIPC-mediated inhibition of TGF-beta signaling[J].Carcinogenesis, 2010, 31(2):175-83.
[15] Kirkbride K C, Townsend T A, Bruinsma M W, et al. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptor[J].JBiolChem, 2008, 283(12):7628-37.
[16] Wiater E, Vale W. Inhibin is an antagonist of bone morphogenetic protein signaling[J].JBiolChem, 2003, 278(10):7934-41.
[17] Chu W, Li X, Li C, et al.TGFBR3, a potential negative regulator of TGF-beta signaling, protects cardiac fibroblasts from hypoxia-induced apoptosis[J].JCellPhysiol, 2011, 226(10):2586-94.
[18] You H J, How T, Blobe G C. The type III transforming growth factor-beta receptor negatively regulates nuclear factor kappa B signaling through its interaction with beta-arrestin2[J].Carcinogenesis, 2009, 30(8):1281-7.
[19] Criswell T L, Arteaga C L. Modulation of NFkappaB activity and E-cadherin by the type Ⅲ transforming growth factor beta receptor regulates cell growth and motility[J].JBiolChem, 2007, 282(44):32491-500.
[20] You H J, Bruinsma M W, How T, et al. The type III TGF-beta receptor signals through both Smad3 and the p38 MAP kinase pathways to contribute to inhibition of cell proliferation[J].Carcinogenesis, 2007, 28(12):2491-500.
[21] Ahn J Y, Park S, Yun Y S, Song J Y. Inhibition of type III TGF-beta receptor aggravates lung fibrotic process[J].BiomedPharmacother, 2010, 64(7):472-6.
[22] Lee N Y, Blobe G C. The interaction of endoglin with beta-arrestin2 regulates transforming growth factor-beta-mediated ERK activation and migration in endothelial cells[J].JBiolChem, 2007, 282(29):21507-17.
[23] Zhang S, Sun W Y, Wu J, J et al. Decreased expression of the type Ⅲ TGF-beta receptor enhances metastasis and invasion in hepatocellullar carcinoma progression[J].OncolRep, 2016, 35(4):2373-81.
[24] Liang H, Zhang C, Ban T, et al. A novel reciprocal loop between microRNA-21 and TGF betaRⅢ is involved in cardiac fibrosis[J].IntJBiochemCellBiol, 2012, 44(12):2152-60.
[25] Hermida N, Lopez B, Gonzalez A, et al. A synthetic peptide from transforming growth factor-beta 1 type Ⅲ receptor prevents myocardial fibrosis in spontaneously hypertensive rats[J].CardiovascRes, 2009, 81(3):601-9.
[26] Schwartze J T, Becker S, Sakkas E, et al. Glucocorticoids recruit Tgfbr3 and Smad1 to shift transforming growth factor-beta signaling from the Tgfbr1/Smad2/3 axis to the Acvrl1/Smad1 axis in lung fibroblasts[J].JBiolChem, 2014, 289(6):3262-75.
[27] Weiner O H, Zoremba M, Gressner A M. Gene expression of syndecans and betaglycan in isolated rat liver cells[J].CellTissueRes, 1996, 285(1):11-6.
[28] Ezquerro I J, Lasarte J J, Dotor J, et al. A synthetic peptide from transforming growth factor beta type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury[J].Cytokine, 2003, 22(1-2):12-20.
[29] Baltanas A, Miguel-Carrasco J L, San J G, et al. A synthetic peptide from transforming growth factor-beta(1) type Ⅲ receptor inhibits NADPH oxidase and prevents oxidative stress in the kidney of spontaneously hypertensive rats[J].AntioxidRedoxSignal, 2013, 19(14):1607-18.
[30] Chen L, Klass C, Woods A. Syndecan-2 regulates transforming growth factor-beta signaling[J].JBiolChem, 2004, 279(16):15715-8.
网络出版时间:2016-12-5 15:14 网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20161205.1514.004.html
Type Ⅲ TGF-β receptor mediated signaling pathway and its role in fibrotic diseases
LI Xin-ran, SUN Wu-yi, GU Yuan-jing, PENG Wen-ting, WEI Wei
(InstituteofClinicalPharmacology,AnhuiMedicalUniversity,KeyLaboratoryofAntiinflammatoryandImmuneMedicine,MinistryofEducation,CollaborativeInnovationCenterofAnti-inflammatoryandImmuneMedicine,Hefei230032,China)
Transforming growth factor β (TGF-β) superfamily ligands play an important role in regulating cellular homeostasis including proliferation, differentiation, apoptosis, immune surveillance and angiogenesis. Type Ⅲ TGF-β receptor (TβRⅢ) is considered to be the coreceptor of TGF-β superfamily. TβRⅢ not only has an effect on classical Smad signaling pathway, but also on non-Smad signaling pathway. TβRⅢ plays a crucial role in fibrosis, tumor, cardiovascular diseases via mediating kinds of signaling pathways. This paper reviews TβRⅢ mediated signaling pathway and its role in fibrotic diseases.
type Ⅲ TGF-β receptor; fibrotic diseases; signaling pathway; β-arrestin2;Smad;MAPK
时间:2016-12-5 15:14
http://www.cnki.net/kcms/detail/34.1086.R.20161205.1514.002.html
2016-07-28,
2016-09-01
国家自然科学基金资助项目(No 81300332,81673444,81330081);安徽省高等学校省级自然科学研究项目(No KJ2012A153)
厉歆然(1990-),女,硕士生,研究方向:肝脏药理学,E-mail:m15555430882@163.com; 孙妩弋(1981-),女,博士,副教授,硕士生导师,研究方向:肝脏药理学,通讯作者,Tel:0551-65161206,E-mail:sunwuyi51@aliyun.com; 魏 伟(1960-),男,博士,教授,博士生导师,研究方向:临床药理学,通讯作者,Tel:0551-65161209,E-mail:wwei@ahmu.edu.cn
10.3969/j.issn.1001-1978.2016.12.001
A
1001-1978(2016)12-1629-06
R-05;R329.25;R349.1;R364.24;R364.32
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
实用肿瘤学杂志(2020年4期)2020-12-08
无机化学学报(2020年7期)2020-07-20
渔业研究(2018年5期)2018-01-17
科技创新导报(2016年30期)2017-03-15
中国卫生标准管理(2015年15期)2016-01-15
中国医疗美容(2015年4期)2015-04-27
郑州大学学报(医学版)(2015年2期)2015-02-27
中国老年学杂志(2015年9期)2015-01-31
中国科技信息(2012年11期)2012-10-26
