
(1R,2S)-2-氟代环丙胺对甲基苯磺酸盐的合成工艺研究
2017-01-05 03:22李学敏
化学工程师 2016年12期
李学敏
(沈阳化工研究院有限公司化工新材料所,辽宁沈阳110021)
(1R,2S)-2-氟代环丙胺对甲基苯磺酸盐的合成工艺研究
李学敏
(沈阳化工研究院有限公司化工新材料所,辽宁沈阳110021)
本文旨在提供一种西他沙星关键中间体的制备方法,其合成路线为:以烯丙基溴为原料,经苄基保护、加成、脱保护、氧化、脱氯、异构体分离、酰氯化、重排得到关键中间体(1R,2S)-2-氟代环丙胺对甲基苯磺酸盐。此工艺具有收率高、操作简便、原料易得、适合工业化生产等优点。
西他沙星;2-氟代环丙胺对甲基苯磺酸盐;合成
西他沙星(Sitafloxacin)[1]是日本第一制药三共株式会社(Daiichi Sankyo)开发的广谱喹诺酮类抗菌药物,分子结构中含有一个顺式氟环丙胺基团,具有良好的药代动力学特性,其体外抗菌活性比大多数同类药物强。此外,此手性氟环丙胺基团还可以减少由于喹诺酮类药物对抗存在前后突触受体的γ-氨基丁酸引起的中枢神经兴奋[2]。由于西他沙星关键中间体(1R,2S)-2-氟代环丙对甲基苯磺酸盐含有两个手性中心,理论上存在4种异构体,其异构体的分离存在很大难度,国内外对其关键中间体的合成研究主要有两种方法:(1)使用二乙基锌与二碘氟甲烷得到氟卡宾,再与烯烃加成[3]等一系列反应后最终得到目标产物,此路线合成步骤长,二乙基锌容易着火且价格非常昂贵,使用时不方便,此外二碘氟甲烷价格昂贵,目前仍无供应商,此路线没有工业化价值;(2)使用重氮乙酸乙酯在催化剂作用下产生卡宾与氟代烯烃反应[4,5],此路线选择性好,但反应条件苛刻,1重氮乙酸乙酯使用过程中容易爆炸,所用催化剂为昂贵的金属铑,这些都限制了其规模化生产。为了找出适于工业化生产的合成工艺,通过对国内外文献报道的查阅对比,本课题组确定采用以烯丙基溴为原料经苄基保护、加成、脱保护、氧化、脱氯、异构体分离与拆分、酰氯化、重排得关键中间体(1R,2S)-2-氟代环丙胺对甲基苯磺酸盐。此路线具有原料易得,产率较高,操作简便等优点,适合工业化生产。
1 实验部分
1.1 试剂与仪器
四氢呋喃、氢化钠、烯丙基溴、NH4Cl、二氯甲烷、NaOH、10%的钯碳、二氯氟甲烷、甲醇、NaClO、NaOH、铝镍合金、乙二胺、浓盐酸、甲基叔丁基醚、异丙醚、乙酸乙酯、草酰氯、叠氮三甲基硅烷,对甲苯磺酸、乙醇、甲苯。
1.2 合成路线与合成方法
1.2.1 合成路线

1.2.2 烯丙基苄醚(A2)的合成
操作:将原料A1溶解到2L四氢呋喃中,开始分批加入NaH,有大量气体放出,加入NaH后保持室温反应5h,不再有气体放出,滴加烯丙基溴,保持温度不超过30℃反应过夜,TLC监测无原料剩余,过滤,滤液用氯化铵淬灭,然后加入1L水,分相、水相用500mL×2四氢呋喃萃取,合并有机相,减蒸馏收集110~120℃(6.0kPa)的馏分,得到色透明液体666g产物A2,收率为97.0%,GC含量98.5%。
1H NMR(500 MHz,CDCl3),ä:7.398~7.309(m,5H),6.056~5.966(m,1H),5.378~5.341(dd,J= 1.52 Hz,J=17.25 Hz,1H),5.253(d,J=10.40 Hz,1H),4.570(s,2H),4.077(d,J=5.56Hz,2H)
1.2.32 -氯-2-氟-1-苄氧甲基环丙烷(A3)的合成
操作:将100gA2溶于2L二氯甲烷中,通入二氯氟甲烷200g,冰浴降温到10℃,在剧烈搅拌下滴入氢氧化钠50%的溶液1L,滴加完毕于0~15℃反应15h。TLC监测有产物生成,GC监测到产物含量85%,停止反应,加入水2L并用二氯甲烷500mL×3萃取,合并有机相,干燥后,减压脱溶得到粗品,然后用油泵蒸出产品70gGC分析92.1%
1H NMR(500 MHz,CDCl3),δ:7.370~7.301(m,5H),4.620~4.504(m,2H),3.719~3.528(m,2H),1.868~ 1.065(m,2H)。
1.2.4 2-氟-2-氯-1-羟甲基环丙烷(A4)的合成
操作:将化合物214gA3加入到3L的单口烧瓶中,然后加入1L甲醇、5.0g钯碳,常温常压加氢反应,反应12h后TLC显示无原料剩余,荧光点消失,过滤,滤液减压脱溶,得到无色液体96.48g,收率98%,GC分析含量97.85%。
1H NMR(500 MHz,CDCl3),δ:3.899~3.535(m,2H),2.268(bs,1H),1.925~0.681(m,3H)。MS,m/Z: 124.01,126.01,125.01
1.2.5 2-氟-2-氯环丙甲酸(A5)的合成
操作:将A4化合物96.48g加入到5L的三口烧瓶中,然后加入乙酸乙酯850mL,NaHCO3150g,于60℃开始滴加10%NaClO溶液,立即放热,滴加完毕后继续搅拌10h,反应液呈无色,GC分析无原料剩余,过滤,酸化水相,并用二氯甲烷3000×3进行萃取,干燥后减压脱溶得到91.26g酸。收率85.3%
1H NMR(500MHz,CDCl3),δ:8.76(s,1H),0.73(m,1H),0.95(m,1H),1.25~1.08(m,1H)。
1.2.6 2-氟环丙甲酸(A6a、A6b、A7a、A7b)的合成
操作:将A5化合物91.26g溶解到630gNaOH 5.7%的溶液中,然后加入铝镍70g合金,于50℃剧烈搅拌,然后同时滴加乙二胺160g和20%NaOH 600g,立即有H2剧烈放出,反应放热,在滴加过程中使得反应温度保持在50~55℃,2h滴加完毕后保温反应3h,反应液呈紫黑色,此时取样GC分析无原料剩余。加入二氯甲烷500mL×3萃取乙二胺,水相用浓HCl酸化,过滤出不溶的乙二胺盐酸盐。并用乙酸乙酯500×4萃取水相,合并有机相然后用无水Na2SO4干燥有机相。减压脱溶得到A6a、A6b、A7a、A7b的混合物。加上精馏柱用油泵减压蒸馏收集40~45℃(2mgHg)馏分得到A6a、A6a,蒸馏釜内剩余的物质为A7a、A7b,乙酸乙酯重结晶得到白色晶体A7a、A7b。30.9g,收率45%,含量98.5%。
1.2.7 Cis-2-氟环丙甲酸A7a的分离
操作:将104gCis-2-氟环丙甲酸(A7a、A7b)加入到1.5L甲苯中使之全部溶解,然后在搅拌下滴入(R)-(+)-N-苄基-1-苯乙胺,加入后于室温下搅拌2h。然后于室温下静置20h,有白色晶体析出。过滤后将晶体加入到0.5L乙酸乙酯中加热回流使之溶解,然后于室温放置20h再次重结晶得到白色固体,按此操作3次,得到晶体94g,将所得道德晶体溶解到1L二氯甲烷中,然后加入20%的NaOH 500mL,并于室温下搅拌0.5h,分出水相,有机相减压脱溶回收B10,水相用浓盐酸酸化到pH=1,然后用乙酸乙酯100mL×4进行萃取,合并有机相减压脱溶得到固体A7a30g收率35.5%。A7a的手性纯度可以将A7a与甲醇反应得到甲酯,然后用带有手型柱的气相色谱进行测定,测定结果为ee=99.6%,。
1H NMR(500 MHz,CDCl3),δ:8.323(bs,1H),4.818(dm,J=63.45 Hz,1H),2.123~1.472(m,2H),1.243~1.071(m,1H)。MS,m/Z:104.1。
1.2.8 (1R,2S)-2-氟代环丙胺对甲基苯磺酸盐A9的合成
操作:于室温下将A7a加入到100mL的单口烧瓶中,然后滴入二氯亚砜,立即有大量的气体生产,滴加完毕后于室温下搅拌过夜,常压蒸出二氯甲烷和未反应的二氯亚砜,然后用水泵减压蒸出产物,收集80℃左右的馏分,得到无色液体34g收率95%。
将此产物和150mL甲苯加入到1L的三口烧瓶中,然后加热到75~80℃,滴加叠氮三甲基硅烷,立即有气体放出,0.5h滴加完毕,于80℃反应3h,此时不再有气体放出,反应完毕冷却至室温。将44g对甲苯磺酸溶解到125mL水中,然后装入500mL的三口烧瓶中,加热到50℃,将上述所得的甲苯溶液滴加到含有对甲苯磺酸的水溶液中,立即有大量的气体生成,0.5h滴加完毕,并于50℃搅拌2h不再有气体放出,冷却到室温分液,将所得的水相减压蒸干得到粘稠的固体,将此固体用乙醇和甲苯混合溶剂重结晶(V/V=1∶2)得到无色鳞片状晶体收率80%。比旋光度[α]20D=-8.33,mp.167~171℃,经过与3,5-二硝基苯甲酰氯衍生化测得产物化学纯度99.9%,光学纯度99.6%。
1H NMR(400 MHz,MeOD),δ:7.71(d,J=8.25 Hz,2.5H),7.25(d,J=8.24 Hz,2.5H),4.83(ddd,J=63. 0,6.5,2.9Hz,2.5H,1H),3.12(m,1H),2.35(s,4H),1.45(m,1H),1.17(m,1H).19FNMR(400MHz,MeOD)δ-215.10。
2 结果与讨论
2.1 合成A3的影响因素
2.2.1 卡宾前体的选择烯丙基苄醚通过与氟卡宾加成反应形成三元环,二碘氟甲烷、二溴氟甲烷、二氯氟甲烷在强碱作用下均可以形成氟卡宾并完成加成反应,虽然3种物质与烯丙基苄醚分别通过单元操作能够制备出西他沙星关键中间体,但二碘氟甲烷没有原料供应商,目前已知的文献用二溴氟甲烷进行闭环反应[6],但二溴氟甲烷价格昂贵,目前仍没有工业化原料。经过对反应机理的深入探讨与分析,我们发现二氯氟甲烷在强碱作用下能形成氯卡宾并与烯丙基苄醚发生加成反应,二氯氟甲烷为制冷剂原料(氟利昂21),价格非常便宜,从工业化角度二氯氟甲烷更适合制备西他沙星中间体。
除了原料成本外,需要考虑的另一个重要因素是两种不同原料闭环产物的收率之间的差别。结果见表1。

表1 反应物料摩尔比对A3收率的影响Tab.1 Effect of molar ratio on the yield of A3
从表1中可以看出,当烯丙基苄基醚的投料量与卡宾前体摩尔比在1∶1~1∶1.1之间时,二溴氟甲烷作为卡宾前体闭环收率高于二氯氟甲烷闭环收率;当烯丙基苄基醚的投料量与卡宾前体摩尔比在1∶1.2~1∶1.3之间时,二溴氟甲烷作为卡宾前体闭环收率与二氯氟甲烷闭环收率基本相同;分析其原因为:二溴氟甲烷常温下为液体沸点66~68℃[7]而二氯氟甲烷物质常温下为气体沸点8.4℃,在反应过程中二氯氟甲烷挥发损失,因此只有当其摩尔比大于1.2时才能有足够的卡宾前体与底物烯丙基苄基醚反应。当继续增加二氯氟甲烷的量,可能由于反应过于剧烈造成副产物增多进而收率下降。经过对比发现:利用二氯氟甲烷替代二溴氟甲烷作为闭环卡宾前体是可行的。
2.2.2 相转移催化剂的选择本反应所用的有机溶剂为二氯甲烷,二氯氟代甲烷溶解其中,在氢氧化的作用下二氯氟代甲烷脱去一个氯原子和一个氢原子形成卡宾,卡宾在与烯丙基苄基醚中的双键发生闭环反应,由于反应过程中是固体氢氧化钠与二氯氟甲烷反的两相应,需要加入相转移催化剂以加快反应进程提高收率,本文考察了相转移催化剂四丁基溴化胺、18-W-6、苄基三乙基氯化铵、四丁基碘化铵对反应收率的影响,其反应结果如下:

表2 相转移催化剂对A3收率的影响Tab.2 Effect of phase transfer catalyst on the yield of A3
从表2可知,在没有催化剂存在下反应收率很低,催化剂能够显著提高收率。在本实验中四丁基碘化铵不仅具有相转移催化作用,其碘原子能够促进二氯氟甲烷中的氯原子离去,因此,四丁基碘化铵催化效果最好。
2.2 合成2-氟环丙烷甲酸的合成与分离
2.2.1 A5的脱氯条件选择已有文献报道A5经过催化脱氯反应[6,8]制备出2-氟环丙甲酸(包括A6a、A6b、A7a、A7b),所用的脱氯还原剂有Pd/C-乙二胺体系、金属钠-乙醇体系、锌粉还原体系、镍-乙二胺还原体系,试验过程中对几种体系进行了对比,其试验结果见表3。

表3 脱氯条件的选择Tab.3 Selection of the conditions for the removal of chlorine
在上表的还原体系中,Pd/C-乙二胺脱氯所需温度和压力均较高,在脱氯的同时有部分三圆环被打开,破坏了三元环结构,造成收率偏低;金属钠乙醇体可在室温常压下脱氯,其氢源来自于钠与乙醇反应产生的H2,由于反应过程中会有乙醇钠生产,特别是反应后期,物料表的粘稠,致使反应不完全,收率偏低;铝镍合金-NaOH体系能够在常温常压下短时间内反应完毕,且收率较高,因此,铝镍合金-NaOH体系作为还原剂最合适。
2.2.2 2-氟环丙甲酸(A6a、A6b、A7a、A7b)异构体的分离由于反应产物2-氟环丙基羧酸具有两个手型中心,因此,有4个异构体,既有顺反异构也有对映异构体,其中A6a、A6b二者为反式异构中的对映异构体,二者有相同的熔点与沸点;A7a、A7b二者为顺式异构中的对应异构体,二者有相同的熔点与沸点。顺式异构与反式异构体之间的沸点差异很大,其中反式结构沸点40~45℃(2mmHg),顺式结构沸点点65~68℃(2mmHg),,因此依靠减压蒸馏的方式将顺反异构分离开来,蒸出的产物为A6a、A6b,蒸馏釜内残余的是A7a、A7b。
A7a、A7b是我们需要的目标产物,因此,本文中主要研究了A7a、A7b的拆分。手型拆分剂(R)-(+)-N-苄基-1-苯乙胺与A7a、A7b形成非对映异构体有机盐,利用非对映异构体的溶解度差别,使其中一个异构体析出,进而完成分离,本实验探讨了不同溶剂对拆分收率的影响,实验结果见表4。
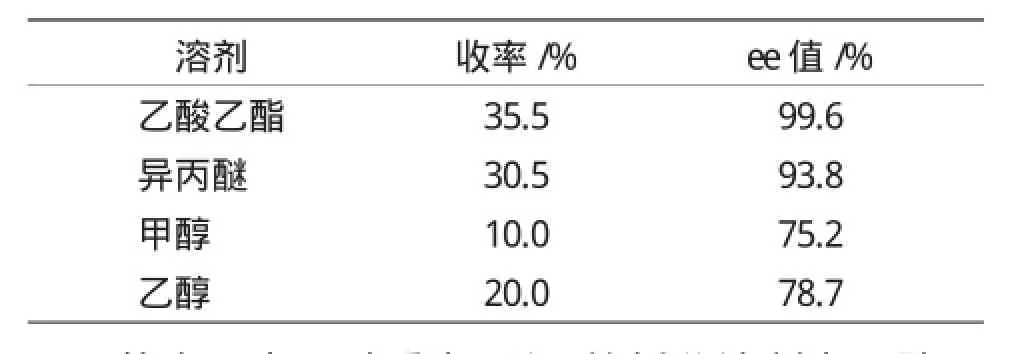
表4 不同溶剂对拆分收率的影响Tab.4 Effect of different solvents on resolution of yield
从表4中可以看出,所用的拆分溶剂为乙酸乙酯时拆分收率和产物光学纯度均较好,分析其原因为甲醇和乙醇极性大,对产物的溶解度大,两种非对映异构体不容易析出,因此,造成拆分收率偏低,此外由于溶剂的极性太大,对非对映异构体的溶解选择性不明显,因此,拆分后产物的光学纯度也不高。乙酸乙酯拆分收率最高,拆分后所得产物的光学纯度也最高,因此,最适合作为拆分溶剂。乙酸乙酯与异丙醚相比,从收率和光学纯度考虑选择乙酸乙酯作为拆分溶剂最合适。
3 结论
(1)以苄醇为原料经过8步反应制备出西他沙星关键中间体(1R,2S)-2-氟代环丙胺对甲基苯磺酸盐,并对各步中间和目标产物进行了结构表征,工艺路线短,各步单元操作简单、原料廉价易得适合工业化生产。
(2)关键中间体总收率为8.87%,化学纯度99.9%光学纯度99.6%,满足制备抗生素原料药要求。
[1]ShohgoA,Masazumi I,Youichi K,et al.Fluorocyclopropylquinolones. Synthesis and structure-activity relationships of 1-(2-fluorocyclopropyl)-3-pyridonecarboxylic acid antibacterialagents[J].Journal ofMedicinal Chemistry,1993,35:3444-3448.
[2]TakahataM,MitsuyamaJ,YamashiroY,et al.Invitroandinvivoantimicrobial activitiesofT-3811ME,anovel des-F(6)-quinolone[J].AntimicrobAgentsChemother,1999,43(5):1077-1084.
[3]陈国彬,岳利剑.合成西他沙星的研究进展[J].合成化学,2012,20(1):7-2.
[4]Toshifumi A,Takanobu I,Hirofumi K,et al.Selectivedehalogenation process[P].US:5780669,1998-07-14
[5]TakuS,Yasumichi F.Stereoselectivesynthesisof cis-2-fluorocyclo propanecarboxylic acid[J].J.Org.Chem.,2014,79:7226-7231.
[6]王印,杜杨.精细化工中间体,2015,32(2):237-240.
[7]Pinaki S B,Meehir P,Mamta S,et al.Fluorinated phosphoniumylides:versatile in situ Wittig intermediates in the synthesis ofhydrofluorocarbons[J].Journal of Fluorine Chemistry,2002,116: 75-80.
[8]Jun-ichi M,Yu-ichirou T.Synthesis ofcis-2-fluorocyclopropylamineby stereoselective cyclopropanation under phase-transfer conditions[J].ChemistryLetters,2004,33:464-465.
[9]Emilie D,Pavel I,Philippe J,et al.Syntheses and applications ofmonofluorinated cyclopropanes[J].Chem.Eur.J.,2012,18:14904-14917.
Study on synthesis process of(1R,2S)-2-fluorocyclopropanamine p-toluenesulfonate
LI Xue-min
(Chemical Advanced Materials Department,Shenyang Research Institute of Chemical Industry,Shenyang 110021,China)
The aim of this paper is to provide one method for the preparation of the key intermediate of sitafloxacin,The synthetic route is:with allyl bromide as raw materials by benzyl protection,addition reaction,deprotection,,oxidation,dechlorination,isomer separation,acylation and rearrangement prepare of the key intermediate(1R,2S)-2-fluorocyclopropanamine p-toluenesulfonate.This process has the advantages of high yield,simple operation,easy to get raw materials,suitable for industrial production,etc.
sitafloxacin;2-fluorocyclopropanamine p-toluenesulfonate;synthesis
TQ073
A
10.16247/j.cnki.23-1171/tq.20161279
2016-10-13
李学敏(1980-),男,工程师,2008年毕业于沈阳化工研究院,硕士研究生学历,应用化学专业,现任专题组长,从事医药农药以及功能染料的开发研究工作。
猜你喜欢
中国调味品(2022年12期)2022-12-05
化工设计通讯(2022年5期)2022-05-25
林产化学与工业(2021年2期)2021-05-11
灾害医学与救援(电子版)(2018年1期)2018-06-05
中国资源综合利用(2017年4期)2018-01-22
国外医药(抗生素分册)(2016年2期)2016-07-12
中国卫生标准管理(2015年4期)2016-01-14
中国塑料(2015年1期)2015-10-14
中国当代医药(2015年36期)2015-03-11
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
