
高效液相色谱法测定复方联苯苄唑乳膏中氢化可的松和联苯苄唑含量
2016-12-29 08:40:00曹月霞
中国药业 2016年23期
曹月霞,蔡 果
(中国医学科学院·北京协和医学院皮肤病医院药剂科,江苏 南京 210042)
高效液相色谱法测定复方联苯苄唑乳膏中氢化可的松和联苯苄唑含量
曹月霞,蔡 果
(中国医学科学院·北京协和医学院皮肤病医院药剂科,江苏 南京 210042)
目的 建立复方联苯苄唑乳膏中氢化可的松和联苯苄唑含量的高效液相色谱(HPLC)法。方法 色谱柱采用Waters Symmetry C18柱(250 mm×4.6 mm,5 μm),流动相为甲醇-水,梯度洗脱,流速为1.0 mL/min,检测波长为242 nm。结果 氢化可的松进样质量浓度在40~200 μg/mL的浓度范围内与峰面积呈良好的线性关系(r=0.999 9),平均回收率为98.48%,RSD为1.07%(n=9);联苯苄唑在40~200 μg/mL的浓度范围内与峰面积呈良好的线性关系(r=0.999 9),平均回收率为99.43%,RSD为0.93%(n=9)。结论 该方法简便准确,重复性好,可用作复方联苯苄唑乳膏中氢化可的松和联苯苄唑的含量控制。
氢化可的松;联苯苄唑;高效液相色普法;含量测定
复方联苯苄唑乳膏是我院自行研制的自制制剂,主要成分为氢化可的松和联苯苄唑。联苯苄唑是一种氮取代、不含卤素的咪唑类局部抗真菌药,抗菌谱广,对皮肤癣菌、酵母菌、曲霉菌、双相真菌均有较强的抑制作用,对某些革兰阳性菌也有抗菌作用[1]。氢化可的松为不含卤素的弱效糖皮质激素,具有抗炎、抗过敏和免疫抑制作用,有显著的抗瘙痒、抗增生作用,用药部位的皮肤萎缩、毛细血管扩张、色素沉着等不良反应较少[2]。氢化可的松弱效激素与抗真菌药的联合应用,可弥补市场众多单方制剂的不足,临床广泛应用于皮肤敏感部位皮炎、湿疹合并真菌感染的治疗,且疗效良好。为提高医院自制制剂质量标准,笔者用高效液相色谱(HPLC)法同时测定两种主药成分含量,方法简便,结果准确,可用于该制剂的质量控制。现报道如下。
1 仪器与试药
1.1 仪器
1525型高效液相色谱系统,2487型紫外检测器,Breeze型色谱数据工作站(美国Waters公司);AE-240型电子分析天平(梅特勒-托利多仪器<上海>有限公司);数显恒温水浴锅(国华电器有限公司)。
1.2 试药
联苯苄唑对照品(中国食品药品检定研究院,批号为100326-201302,纯度为98.6%);氢化可的松对照品(中国食品药品检定研究院,批号为 100152-200206,纯度为100%);复方联苯苄唑乳膏(自制制剂,批号为20160114,20160527,20160616);辅料均为药用规格,甲醇为色谱纯,水为重蒸水。
2 方法与结果
2.1 色谱条件
色谱柱:Waters Symmetry C18柱(250 mm×4.6 mm,5 μm);流动相:甲醇(A)-水(B),梯度洗脱,见表1;流速:1.0 mL/min;检测波长:242 nm;柱温:35℃;进样量:10 μL。

表1 流动相梯度洗脱表
2.2 溶液制备
对照品溶液:称取氢化可的松、联苯苄唑对照品各10.00 mg,精密称定,置100 mL容量瓶中,加甲醇适量使溶解,并稀释至刻度,即得质量浓度为100 μg/mL的氢化可的松和100 μg/mL的联苯苄唑对照品溶液。
供试品溶液:取供试品适量(相当于联苯苄唑和氢化可的松各10 mg),精密称定,置于烧杯中,加甲醇约20 mL,水浴加热,搅拌使其溶解,全部转移至100 mL容量瓶中,冷却至室温加甲醇稀释至刻度,摇匀,即得。
阴性对照品溶液:按处方比例配置不含联苯苄唑和氢化可的松的阴性样品,按供试品溶液制备方法制得。
2.3 方法学考察
系统适用性试验:按拟订色谱条件,分别进样对照品溶液,并记录色谱图见图1。结果表明,供试品溶液色谱图中,氢化可的松峰与联苯苄唑峰分离度良好,保留时间分别为10.3 min,26.6 min,理论板数分别为2 750,800,峰型良好。
空白干扰试验:按拟订色谱条件,进样阴性样品溶液,结果显示在氢化可的松和联苯苄唑对照品溶液色谱峰处无干扰峰,表明制剂中其他药用辅料成分对氢化可的松和联苯苄唑的测定无干扰(见图1)。
线性关系考察:精密称取氢化可的松对照品10 mg,置50 mL容量瓶中,加甲醇溶解并定容至刻度,摇匀,即得200 μg/mL贮备液;精密吸取该溶液2.0,4.0,6.0,8.0 mL,分别置10 mL容量瓶中,用甲醇稀释至刻度,摇匀,取上述溶液及贮备液按上述色谱条件进样,记录色谱图。以氢化可的松质量浓度(C)为横坐标、平均峰面积(A)为纵坐标进行线性回归,回归方程为 A=21 047C+43 250,r=0.999 9(n=5)。结果表明,氢化可的松质量浓度在40~200 μg/mL范围内与峰面积呈良好的线性关系。精密称取联苯苄唑对照品10 mg,置50 mL容量瓶中,加甲醇溶解并定容至刻度,摇匀,即得200 μg/mL贮备液;精密吸取该溶液2.0,4.0,6.0,8.0 mL,分别置10 mL容量瓶中,用甲醇稀释至刻度,摇匀,取上述溶液及贮备液按上述色谱条件进样,记录色谱图。以联苯苄唑的质量浓度(C)为横坐标、平均峰面积(A)为纵坐标进行线性回归,回归方程为 A=41 709C+90 829,r=0.999 9(n=5)。结果表明,联苯苄唑质量浓度在40~200 μg/mL范围内与峰面积呈良好的线性关系。
精密度试验:取同一对照品溶液,重复进样5次,记录色谱图。结果氢化可的松和联苯苄唑的峰面积 RSD分别为1.54%(n=5)和1.09%(n=5),表明仪器精密度良好。
稳定性试验:取同一供试品溶液适量,于室温下放置0,2,4,6,8 h,进样并记录色谱图。结果氢化可的松和联苯苄唑的峰面积 RSD分别为1.56%和1.44%(n=5)。表明供试品溶液在8 h内稳定。
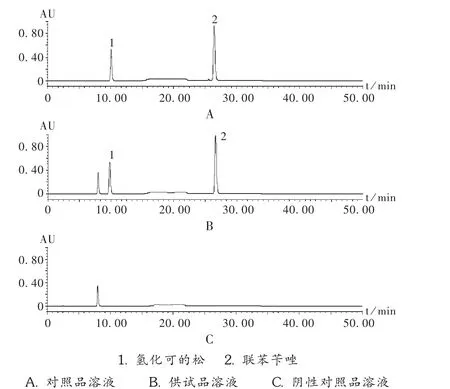
图1 高效液相色谱图
重复性试验:取同一批样品适量,共6份,按供试品溶液制备方法制得供试品溶液,按拟订色谱条件测定,结果氢化可的松和联苯苄唑的含量 RSD分别为1.47%和1.37%(n=6),表明该方法重复性良好。
加样回收试验:分别按照样品标示量的 80%,100%,120%,精密称取氢化可的松和联苯苄唑原料药,按处方比例制得样品,按供试品溶液制备方法制备高、中、低不同浓度的供试品溶液,各3份,照拟订色谱条件测定。结果见表2。
2.4 样品含量测定
取不同批号样品,按外标法以峰面积计算其含量。结果见表3。
3 讨论
复方联苯苄唑乳膏为我院自制制剂,由于处方中基质与药物互相干扰,很难以常规检测方法同时对两种主药成分进行含量测定,根据《中国药典》方法及文献[3-9]的含量测定方法,或只能测得单一成分的含量,或不能排除基质成分对试验结果的影响,两主药及基质成分不能有效分离,不能达到同时检测的目的。文献[10]虽能用薄层色谱法鉴别复方联苯苄唑乳膏中两种主药成分,但对其含量不能确认。
提取条件选择:参考文献[11]“加无水乙醇30 mL,在水浴上加热使溶解,再置冰浴中冷却,滤过。如此提取3次,滤液并人100 mL烧杯中,于水浴上挥干用流动相溶解,放冰箱中冷藏过夜,迅速过滤,滤液转移至100 mL容量瓶中并稀释至刻度,摇匀,即得”。提取时间更短,操作更加简便,更适合用于医疗机构自制制剂的快速检测。对供试品的提取次数进行比较,用甲醇分别进行1,2,3次提取,取供试液进样。结果表明,提取次数对结果影响甚微,从节约成本角度考虑,选择甲醇提取1次。

表2 复方联苯苄唑乳膏加样回收试验结果(n=9)
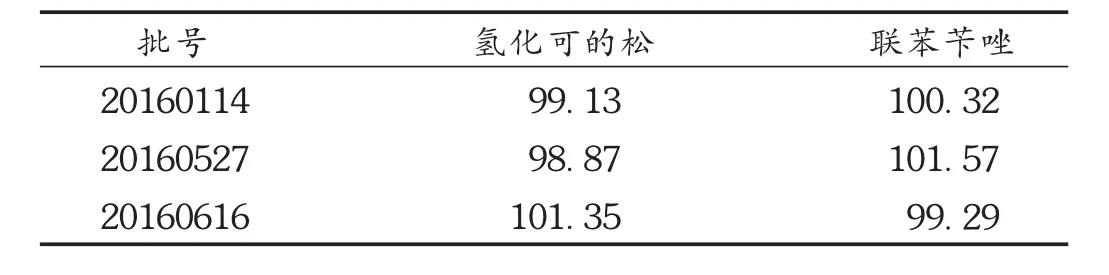
表3 复方联苯苄唑乳膏含量测定结果(%,n=3)
波长选择:根据《中国药典(二部)》方法,氢化可的松的最大吸收波长为 242 nm[12]766,联苯苄唑的最大吸收波长为 254 nm[12]1263。由于氢化可的松于 254 nm波长处吸收较小,联苯苄唑于242 nm波长处吸收较大,故选择242 nm作为测定波长,两种主药成分峰面积均较大,检测灵敏度较高。
流动相选择:参考文献[13],所选用的流动相为磷酸盐缓冲液-乙腈-甲醇(25∶15∶60)等度洗脱,或水相-甲醇(醋酸铵26 g和三乙胺37 mL,加水至1 000 mL,冰醋酸调pH至4.0)(45∶55)为流动相[14]。本试验中对流动相的比例进行考察,试验考察了78%,80%,82%,85%比例的甲醇对试验结果的影响,结果显示,甲醇比例的变化对联苯苄唑保留时间影响较大,比例越大,则联苯苄唑的保留时间越短;然而比例过大,则各组有效成分不能有效分离,且梯度变化使基线较不平稳。故而最终选择80%比例的甲醇,梯度洗脱,基质与两主药成分可有效分离,主药保留时间也较为合适。
综上所述,该试验所用方法充分考虑了制剂制备工艺和两主药成分的性质,优化了实验操作步骤,可有效排除基质峰对两主药色谱峰的影响,等度不能把基质和两组分有效地分离,最终选用本法进行梯度洗脱,最终达到满意的分离效果。所建立的方法操作简便,节约成本,经济效率,可作为本医疗机构该制剂快速检测的方法。
[1]王 斌,王宏图.局部抗真菌药联苯苄唑[J].中国新药与临床杂志,1999,18(4):243-245.
[2]张建中.几种新的糖皮质激素[J].中国药物应用与检测,2005,2(5):30-33.
[3]杨 帆,白 林.氢化可的松含量测定方法改进[J].中国药业,2011,20(15):25.
[4]江 生,张晓松.高效液相色谱法测定联苯苄唑凝胶的含量[J].中国药业,2005,14(5):34-35.
[5]刘艳娥,苗惠珠,刘玉波.高效液相色谱法测定联苯苄唑乳膏中联苯苄唑的含量[J].解放军药学学报,2001,17(1): 44-46.
[6]邵建兵.氢化可的松洗剂及霜剂的含量测定[J].泰州职业技术学院学报,2010,10(3):36-37.
[7]钱燕媚,伍名兰,郑 婕.HPLC法测定不同厂家联苯苄唑凝胶的含量[J].中国药房,2016,27(15):2 117-2 119.
[8]张天虹,徐 文,崔升淼,等.高效液相色谱法测定联苯苄唑凝胶中联苯苄唑的含量[J].实用药物与临床,2003,6(4):174-175.
[9]单萍萍,申国庆.高效液相色谱法测定复方联苯苄唑搽剂中联苯苄唑和氢化可的松的含量[J].江苏药学与临床研究,2006,14(1):26-28.
[10]于明彦.薄层色谱法鉴别复方联苯苄唑乳膏和洗剂中联苯苄唑与氢化可的松[J].药物分析杂志,2000,20(6):429-430.
[11]宁德英.高效液相色谱法测定复方氢化可的松霜中氢化可的松的含量[J].时珍国医国药,2005,19(2):11-12.
[12]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2015.
[13]王 艳,徐 晖,刘玉强,等.HPLC法测定乳膏中联苯苄唑和醋酸曲安奈德的含量[J].中国新药杂志,2007,16(8): 631-633.
[14]徐英宏,杨 君.HPLC同时测定氢定乳膏中氢化可的松和醋酸氯己定的含量[J].中国药学杂志,2006,41(15):1 184-1 186.
Content Determination of Hydrocortisone and Bifonazole in Compound Bifonazole Cream by HPLC
Cao Yuexia,Cai Guo
(Chinese Academy of Medical Sciences·Peking Union Medical College Dermatological Hospital,Nangjing,Jiangsu,China 210042)
Objective To establish an effective HPLC method for the content determination of hydrocortisone and bifonazole in Compound Bifonazole Cream.M ethods The separation was performed on Waters Symmetry C18(250 mm×4.6 mm,5 μm)with different proportions of methanol-water at a flow rate of 1.0 mL/min with gradient elution,and UV detection was set at 242 nm.Results A good linearity was obtained over the range of 40-200 μg/mL(r=0.999 9)for hydrocortisone and 40-200 μg/mL(r=0.999 9)for bifonazole,respectively.The average recovery rates of these two components were 98.48% with RSD=1.07%(n=9)and 99.43% with RSD=0.93%(n=9).Conclusion The method is simple,accurate and reproducible,which can be used for the content control of the preparation.
hydrocortisone;bifonazole;HPLC;content determination
R927.2;R986
A
1006-4931(2016)23-0065-03
曹月霞(1992-),女,江苏南京人,药师,主要从事医院药剂科工作,(电子信箱)1525972154@qq.com。
2016-08-01;
2016-09-24)
猜你喜欢
中西医结合心血管病电子杂志(2020年5期)2020-06-08 15:49:48
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
现代临床医学(2016年2期)2016-02-19 17:51:51
合成化学(2015年2期)2016-01-17 09:03:13
化工进展(2015年3期)2015-11-11 09:08:25
橡胶工业(2015年2期)2015-07-29 08:29:46
化学分析计量(2015年4期)2015-03-23 16:47:34
西南军医(2014年5期)2014-04-25 07:42:49
食品工业科技(2014年9期)2014-03-11 18:15:39
