
高通量全基因组测序法对尿道致病性大肠埃希菌NB8株毒力因子数据的初步分析
2016-12-14 03:15翁幸鐾糜祖煌王春新朱健铭
温州医科大学学报 2016年11期
翁幸鐾,糜祖煌,王春新,朱健铭
(1.宁波市第一医院 检验科,浙江 宁波 315010;2.无锡市克隆遗传技术研究所 生物信息学室,江苏无锡 214026;3.南京医科大学附属无锡人民医院 检验科,江苏 无锡 214023;4.杭州市余杭区中医院 检验科,浙江 杭州 311106)
高通量全基因组测序法对尿道致病性大肠埃希菌NB8株毒力因子数据的初步分析
翁幸鐾1,糜祖煌2,王春新3,朱健铭4
(1.宁波市第一医院 检验科,浙江 宁波 315010;2.无锡市克隆遗传技术研究所 生物信息学室,江苏无锡 214026;3.南京医科大学附属无锡人民医院 检验科,江苏 无锡 214023;4.杭州市余杭区中医院 检验科,浙江 杭州 311106)
目的:调查1株尿道致病性大肠埃希菌(UPEC)NB8株携带的毒力因子和可能存在的致病机制。方法:UPEC NB8株分离自2012年4月宁波市第一医院住院患者尿液标本,做16SrDNA和gyrA基因测序BLASTn比对确认为UPEC,用Illumina HiSeq与Ion Torrent PGM这2种大规模并行测序仪进行全基因组分析,再进行人工测序补缺口。再对NB8株做毒力因子数据挖掘和分析,最后把NB8株和9株全基因组测序的UPEC的毒力因子做比较基因组学研究。结果:UPEC NB8株全基因组测序得到一条推定的染色体序列,长4 550 369 bp(内含14个缺口)。推定的质粒序列,长635 377 bp(内含33个缺口)。NB8株全基因组中挖掘出黏附(黏附素、P菌毛、1型菌毛、大肠埃希菌共同菌毛)、铁摄取系统(产气菌素、血红素摄取、肠杆菌素)、蛋白酶(分泌自转运毒素)、毒素(溶血素)、抗原43、SHI-2毒力岛等多种毒力因子。结论:UPEC NB8株中检出的多种毒力因子可能会在细菌黏附、侵袭、溶细胞活性、细胞毒性等方面起着重要作用,并能增强细菌的生存能力和致病力,导致宿主的泌尿道感染。全基因组测序技术具有超高速、高通量、低成本和高效益的优势,值得推广。
大肠埃希菌;全基因组;多药耐药;尿道致病性;毒力因子
每年超过800万美国女性受尿道致病性大肠埃希菌(uropathogenic Escherichia Coli,UPEC)感染而产生泌尿道感染(urianary tract infaction,UTI)症状[1],33%女性在24岁时已有UTI史并接受抗生素治疗,超过半数的女性在一生中曾有过UTI史[2],大于80%的UTI由UPEC引起[3]。急性UTI可致急性膀胱炎和急性肾盂肾炎,最终甚至可致肾衰竭、尿毒症。国内对UPEC的研究较少,我们曾报道了用PCR法对1组28株多耐药UPEC做了β-内酰胺类[4-5]、氨基糖苷类[6]、喹诺酮类耐药基因[7]、抗菌制剂外排泵基因[8]、可移动遗传元件遗传标记[9]介导的分子耐药机制研究和几种毒力因子[10]介导的分子致病机制研究。为了更深入了解UPEC的遗传学背景,本研究分别采用了Illumina HiSeq与Ion Torrent PGM这2种大规模并行测序仪对1株多耐药UPEC NB8株进行全基因组分析,并从全基因组序列中挖掘了多种毒力因子,现报告如下。
1 材料和方法
1.1菌株来源 UPEC NB8株分离自2012年4月宁波市第一医院高干病房1名83岁女性住院患者的尿液标本。该患者因“尿路感染”住院,立即予以哌拉西林/他唑巴坦进行治疗,但患者病情迅速恶化并死亡。该菌株先用法国生物梅里埃公司VITEK 2-Compact全自动微生物鉴定仪及配套革兰阴性菌鉴定卡进行鉴定,再作16SrDNA和gyrA基因测序,经GenBank比对进一步确认为UPEC。
1.2药物敏感性试验 采用法国生物梅里埃公司VITEK 2-Compact全自动微生物鉴定仪及配套革兰阴性菌药敏卡进行16种药物敏感性检测;红霉素、四环素、美罗培南的药敏试验采用纸片扩散法,M-H琼脂为法国生物梅里埃公司产品,药敏纸片为英国Oxoid公司产品。再根据2012年美国临床实验室标准化研究所(CLSI)的标准进行抗菌药物敏感性判断。NB8株对氨苄西林(≥32)、头孢唑啉(≥64)、头孢曲松(≥64)、头孢他啶(≥64)、头孢吡肟(≥64)、氨曲南(≥64)、头孢替坦(≥64)、氨苄西林/舒巴坦(≥32)、庆大霉素(≥16)、妥布霉素(≥16)、丁胺卡那(≥64)、环丙沙星(≥4)、左氧氟沙星(≥8)、复方新诺明(≥320)、红霉素(7 mm)、四环素(7 mm)均耐药;对哌拉西林/他唑巴坦(64)、头孢哌酮/舒巴坦(17 mm)中介;对亚胺培南(≤1)、美罗培南(27 mm)敏感。
1.3全基因组测序
1.3.1测序策略:样本DNA采取随机散弹式打断来构建DNA文库,并使用Illumina HiSeq与Ion Torrent PGM这2种平台进行测序。组装之后再利用PCR等方法进行补缺口。
1.3.2测序流程:在Illumina HiSeq平台的测序流程,取1 μg的genomic DNA,利用Nebulization方法将DNA打断成约800 bp的片段,纯化后接上PE adaptors然后进行双向定序,得到序列数据。在Ion Torrent PGM平台的测序流程,取100 ng的genomic DNA,利用Ultrasonication方法将DNA打断成约400 bp的片段,纯化后接上adaptor。然后个别与磁珠上的adaptor杂合并进行扩增,最后在314半导体芯片上进行定序反应,得到序列数据。
1.3.3组装结果:使用软件将clean reads先进行de novo组装后,得到的contigs除了利用paired-end information得到scaffolds之外,再与参考基因组:大肠埃希菌SE15(Accession number:NC_013654)进行对照与排列顺序,可以得到1条推定的染色体(有缺口)。全基因组测序委托中国台湾基龙米克斯生物科技股份有限公司进行。
1.3.4补缺口:针对缺口设计引物,得到PCR产物后再使用Sanger测序法,将大部分缺口补起来。
1.4生物信息分析
1.4.1基因注释:使用Glimmer v3.02与prokaryotic GeneMark.hmm v2.10f对推定的染色体与contigs进行ORF预测(Start codons使用ATG & GTT;Stop codons使用TAG & TGA & TAA),并利用RBSfinder程序找出基因的转录起始点附近的核糖体连接位点。使用RNAmmer(v1.2)与tRNAscan-SE(v1.23),进行rRNA & tRNA基因的预测。
1.4.2功能注释:将预测出的基因段落转成蛋白质序列分别与NCBI的nr & microbial RefSeq protein databases & COG databases以及KEGG的protein database进行BLASTP比对,得到基因注释。
1.5毒力因子比较基因组学研究 把NB8株和9株完成全基因组测序的UPEC做毒力因子的比较基因组学研究。
2 结果
2.1全基因组组装结果 UPEC NB8株经Illumina HiSeq与Ion Torrent PGM测序仪进行平行测序,2种仪器测得序列合并组装后PCR法补缺口。最终得到1条推定的染色体序列,长4 550 369 bp(内含14个缺口)。推定的质粒序列,长635 377 bp(内含33个缺口)。
2.2全基因组功能注释结果 COG基因功能分类提示NB8株的功能基因共有3 922个,按功能分为3类:代谢类基因最多,共1 643个(占41.89%),其中糖类转运和代谢基因371个(占9.46%),氨基酸转运和代谢基因359个(占9.15%),产能和储能基因275个(占7.01%),无机铁转运和代谢基因245个(占6.25%),辅酶转运和代谢基因147个(占3.75%);第2类为信息储存和处理基因,共629个(占16.03%),其中转录相关基因242个(占6.17%),复制、重组和修复基因208个(占5.30%),翻译、染色体结构和起源基因177个(占4.51%);第3类为细胞过程和信号转导基因,共860个(占21.92%),其中细胞壁/膜/外壳起源基因239个(占6.09%),翻译修饰、蛋白翻转、分子伴侣基因基因142个(占3.62%),信号转导基因137个(占3.49%),细胞内运输,分泌和小泡运输基因129个(占3.29%),细胞运动基因122个(占3.11%)。另有功能预测基因460个,功能未知基因330个。用KEGG反应代谢通路分析也得到类似的结果。同源蛋白来源物种表提示NB8株来源于大肠埃希菌的蛋白有5 003个,占97.5%,还有少量的蛋白可能来源于肺炎克雷伯菌、沙门菌和志贺菌等,需要做进一步的群体遗传学分析。NB8株全基因组中挖掘出黏附(黏附素、P菌毛、1型菌毛、大肠埃希菌共同菌毛)、铁摄取系统[产气菌素、血红素摄取(Chu)、肠杆菌素]、蛋白酶[分泌自转运毒素(Sat)]、毒素(溶血素)、抗原43、SHI-2毒力岛等多种毒力因子,见表1。对于细菌分泌系统通路,NB8株参与I、 II、 IV、V 4种分泌系统通路,主要参与 II型分泌系统,见图1。NB8株携带的FliD与菌毛帽相关,FlgL和FlgK与钩-菌毛结合相关,FlgF、FlgB、FlgC与近端杆相关,FlgG与远端杆相关,FlgH与L环相关,FlgI与P环相关,FliF与MS环相关,FliG、FliN与C环相关,FlhA、FlhB与鞭毛输出装置相关,FliI、FliP与 III型分泌系统相关,MotA、MotB与鞭毛马达相关,见图2。
将NB8和NCBI中9株已完成全基因组测序的UPEC,共10株菌做了毒力因子比较基因组学研究,发现这10株菌都有黏附、自转运蛋白、侵袭、铁摄取、毒素等多种毒力因子,但NB8株携带的菌毛基因和ACE T6SS较少,铁摄取基因非常丰富,见表2。

表1 UPEC NB8株全基因组测序后毒力因子挖掘

续表1
3 讨论
本研究中的NB8株因产生高毒力,导致患者病情迅速恶化后死亡,因此,挖掘并分析NB8株携带的多种毒力因子就有重要的研究价值。UPEC致病性强、黏附力强、难以清除,使得泌尿道感染病情反复发作、迁延不愈,其中菌毛在黏附、侵袭等方面起着重要作用。常见的菌毛主要包括1型菌毛和P菌毛,本研究均有检出。其中1型菌毛最常见,90%的菌株均有携带。由fim基因编码的Fim蛋白位于1型菌毛的顶端,可以识别泌尿道上皮的甘露糖受体,并与之结合,使大肠埃希菌吸附于膀胱上皮细胞起关键作用[12],避免因震荡、冲刷而脱离黏膜上皮,为进一步侵入机体创造条件。pap基因编码的P菌毛是UPEC黏附定植的关键因子,有助于细菌与上尿路泌尿道上皮细胞的黏附并能直接介导炎症反应[13]。

图1 UPEC NB8株全基因组中细菌分泌系统通路图
除了菌毛,UPEC的致病性还取决于从宿主体内竞争铁的能力,UPEC在环境中获得所需的铁,能增强其生存能力和致病力。一种直接的摄铁方式是从血红素或含血红素蛋白中竞争铁离子,UPEC血红素的利用是由chu基因簇(ccmA、B、C、D基因)编码一系列蛋白,参与转运血红素到细菌细胞内还有后续的加工处理。第二种是间接的摄铁机制,是利用铁结合复合物,它们能高亲和性地结合铁离子。第三种摄铁机制是分泌一些低分子量铁载体,将铁转运至细胞内。fep是肠杆菌素的转运基因,本研究检出的fepC为ATP结合蛋白,编码高铁肠杆菌素转运ATP结合蛋白,该基因簇与铁载体—肠杆菌素的转运有关。iuc基因簇(iucA、iucB、iucC)和铁载体相关,其中iucA和含铁产气菌素一个特定的外膜受体蛋白的表达相关,iucB和产气菌素的合成相关[14]。上述3种摄铁方式在本研究中均有检出,提示NB8株有强大的摄取铁能力来增强其致病力。分泌自转运毒素(Sat)对膀胱和肾脏来源的细胞具有毒性:能引起肾脏的严重损伤,导致肾小球膜的溶解,肾小管上皮细胞的操作和肾脏组织的空泡化[15]。本研究检出3种丝氨酸蛋白酶自转运体基因,50% UPEC能检出hlyA编码的α-溶血素,HlyD属于膜融合蛋白,是α-溶血素分泌途径特异性通道蛋白。成熟的α-溶血素进入宿主细胞膜,使细胞内容物释放,具有溶细胞活性和细胞毒性[16]。
抗原43是大肠埃希菌表达的一种表面抗原蛋白分子,具有强大的免疫原性,含有多种T细胞和B细胞表位[17]。SHI-2毒力岛基因来源于志贺氏菌,位于selC基因下游,主要介导铁结合蛋白产气菌素的合成和运输[18],本研究检出双拷贝SHI-2毒力岛基因。
把NB8株和NCBI中9株已完成全基因组测序的UPEC,共10株菌做了毒力因子比较基因组学研究,发现这10株菌都有黏附、自转运蛋白、侵袭、铁摄取、毒素等多种毒力因子,但NB8株携带的菌毛基因和ACE T6SS较少,提示NB8株人黏附能力和细菌分泌功能较弱,需要进一步做功能验证。而铁摄取基因非常丰富,提示NB8株有强大的摄取铁能力来增强其致病力。
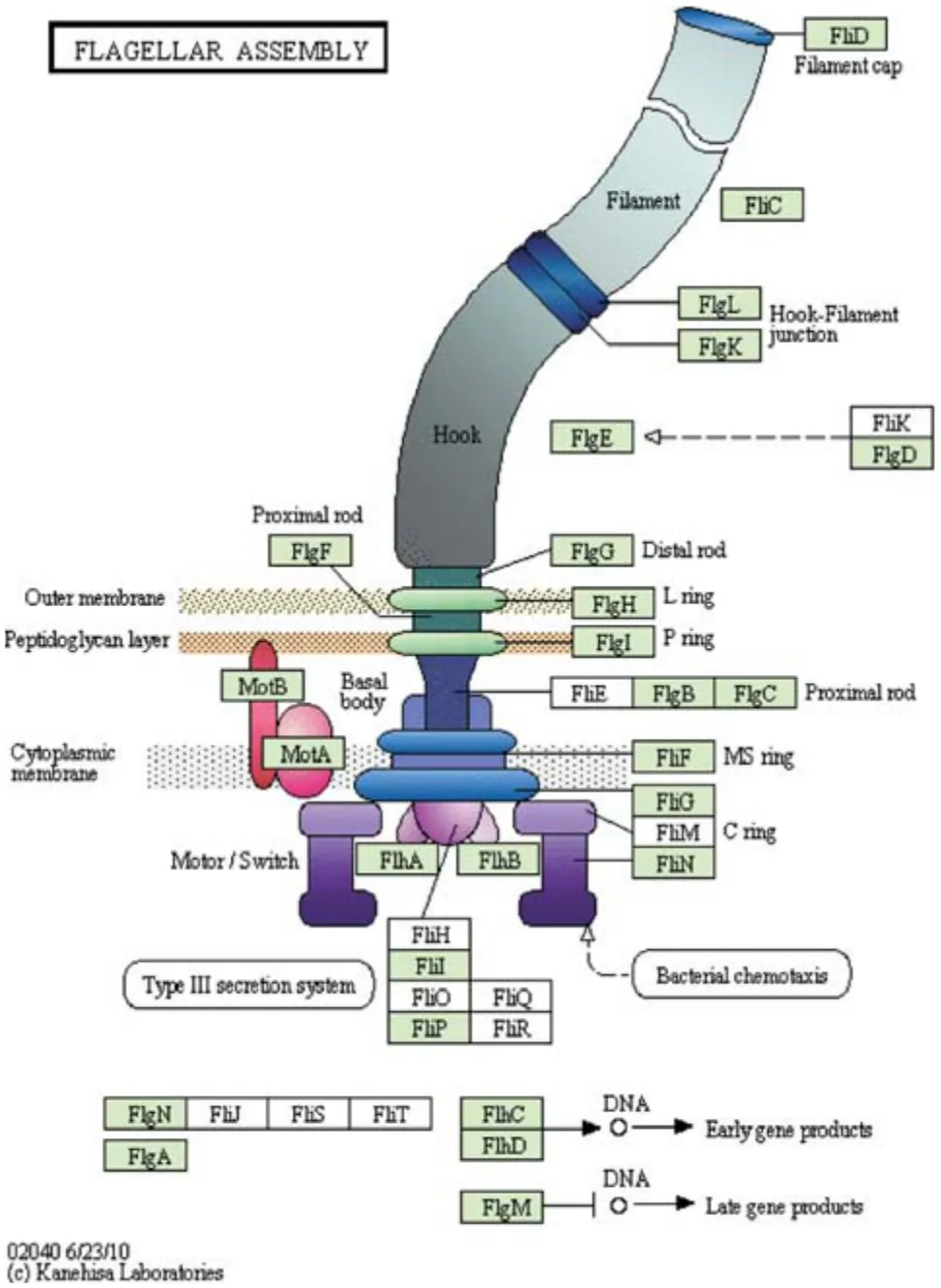
图2 NB8株全基因组中鞭毛装配通路图

表2 10株UPEC全基因组测序后毒力因子比较基因组学研究

续表2
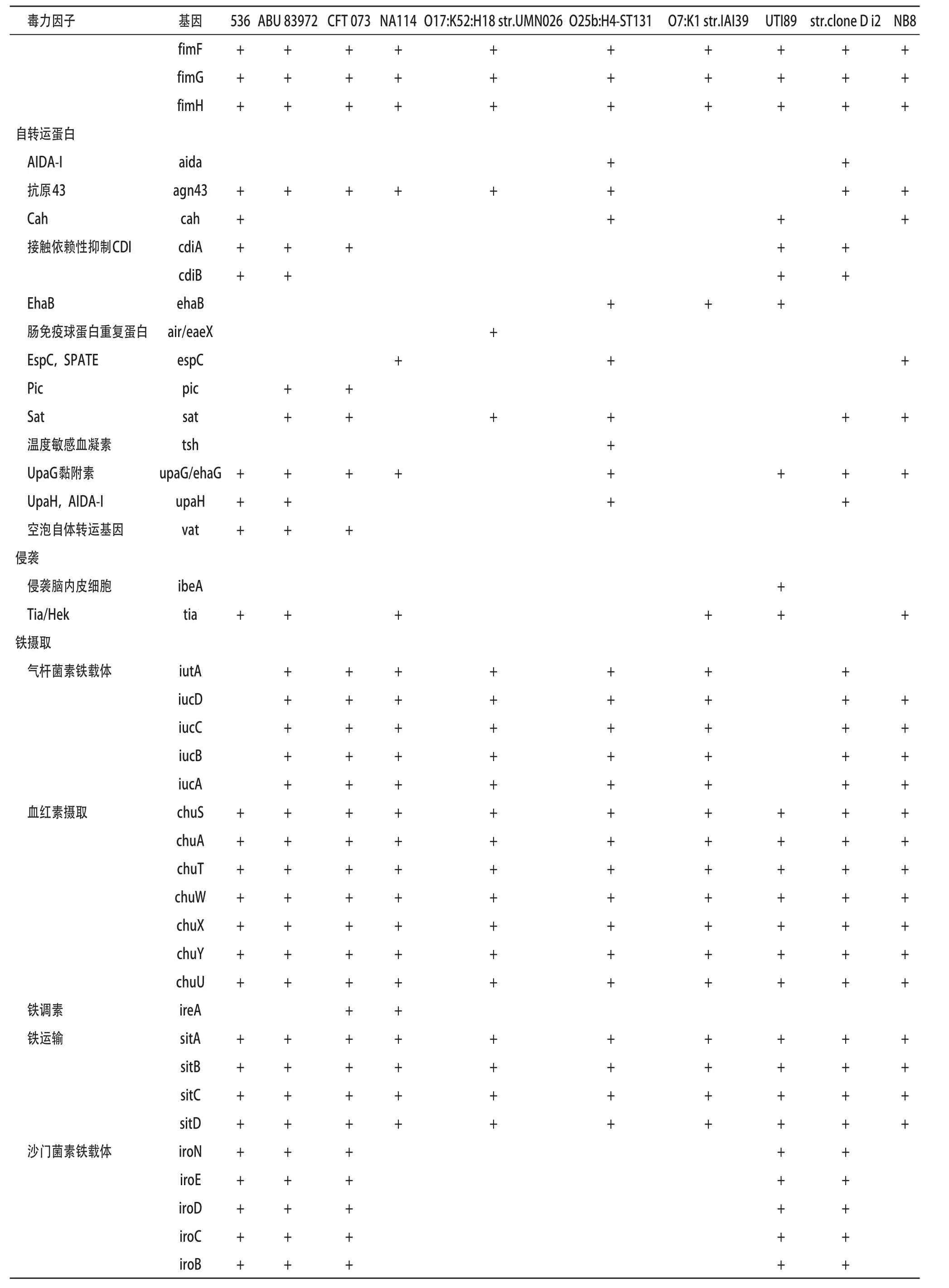
续表2

续表2
与传统的Sanger测序技术相比,新一代测序(next-generation sequencing,NGS)技术具有超高速、高通量、低成本、高效益、无需设计引物、能检出基因的多拷贝和新基因等优势。NGS技术已经用在细菌毒力因子挖掘的研究中[19]。现有NGS技术缺憾为测序仪读序列(reads)长度较短,通常会包含错误。当基因组中出现大量短的重复序列时即无法正准拼接序列,不能测通全长染色体而出现缺口。NB8株经Illumina HiSeq与Ion Torrent PGM仪器测得序列合并组装后PCR法补缺口,最终得到推定的染色体序列中有14个contig之间的顺序及正反关系均未知,因此含14个缺口,同理推定的质粒序列含33个缺口。全基因组测序理论上可测出细菌的全部染色体和质粒序列,可读出全部功能基因,包括耐药基因和临床医师深度关切的与细菌致病性相关的毒力基因,故全基因组分析在国内外已逐步用在细菌的耐药机制和致病机制,以及耐药菌传播途径分析中[20-21]。
本研究对UPEC NB8株毒力因子进行了初步的生物信息学分析,后续会进一步做毒力因子的功能验证,以明确毒力因子的机制和致病机制。
[1] BUSH L M, CHAPARRO-ROJAS F, OKEH V, et al. Cumulative clinical experience from over a decade of use of levofl oxacin in urinary tract infections: critical appraisal and role in therapy[J]. Infect Drug Resist, 2011, 4: 177-189.
[2] FIHN S. Acute uncomplicated urinary tract infection in Women [J]. N Engl J Med 2003, 349(3): 259-266.
[3] FOXMAN B. The epidemiology of urinary tract infection[J]. Nat Rev Urol, 2010, 7(12): 653-660.
[4] 翁幸鐾, 糜祖煌. 大肠埃希菌尿液分离株β-内酰胺酶基因、AmpC酶基因和膜孔蛋白ompC基因研究[J]. 中华微生物学和免疫学杂志, 2010, 30(12): 1083-1084.
[5] 翁幸鐾, 糜祖煌. 大肠埃希菌对β-内酰胺类药物耐药特征及分子流行病学研究[J]. 国际流行病学传染病学杂志, 2011, 38(6): 375-380.
[6] 翁幸鐾, 糜祖煌. 从尿液中分离出aac(6’)-Ib基因新亚型的大肠埃希菌[J]. 中华检验医学杂志, 2010, 33(2): 149-151.
[7] 翁幸鐾, 糜祖煌. 大肠埃希菌尿液分离株发现gyrA基因新变异株[J]. 中华微生物学和免疫学杂志, 2010, 30(1): 11-16.
[8] 翁幸鐾, 糜祖煌. 大肠埃希菌尿液分离株7种抗菌制剂外排泵基因研究[J]. 中华医院感染学杂志, 2010, 20(6): 759-762.
[9] 翁幸鐾, 糜祖煌. 大肠埃希菌尿液分离株可移动遗传元件研究[J]. 中华医院感染学杂志, 2010, 20(5): 607-610.
[10] 翁幸鐾, 糜家睿. 多耐药大肠埃希菌尿液分离株毒力基因检出ompT基因新亚型[J]. 中华检验医学杂志, 2012, 35(2): 176-179.
[11] CLSI. Performance standards for antimicrobial susceptibility testing[M]. 22nd Informational Supplement, 2012.
[12] FLOYD K A, MOORE J L, EBERLY A R, et al. Adhesive fiber stratification in uropathogenic Escherichia coli biofilms unveils oxygen-mediated control of type 1 pili[J]. PLoS Pathog, 2015, 11(3): e1004697.
[13] LANE M C, MOBLEY H L. Role of P fi mbria1 mediated adherence in pyelonephritis andpersistenee of uropathogenic Escherichia coil (UPEC) in the mammalian kidney[J]. Kidney Int, 2007, 72(1): 19-25.
[14] XIONG L, LING J, GAO Q, et al. Construction of iucB and iucBiutA mutants of avian pathogenic Escherichia coli and evaluation of their pathogenicity[J]. Vet Microbiol, 2012, 159(3-4): 420-431.
[15] NICOLAS V, LIÉVIN-LE MOAL V. Antisecretory factor peptide AF-16 inhibits the secreted autotransporter toxinstimulated transcellular and paracellular passages of fl uid in cultured human enterocyte-like cells[J]. Infect Immun, 2015, 83(3): 907-922.
[16] 宿玲恰, 陈晟, 吴敬. 大肠杆菌α-溶血素分泌途径研究进展[J]. 微生物学报, 2013, 53(10): 1011-1017.
[17] KAMINSKA R, VAN DER WOUDE M W. Establishing and maintaining sequestration of dam target sites for phase Variation of aga43 in Escherichia coli[J]. J Bacteriol, 2010, 192(7): l937-1945.
[18] VOKES S A, REEVES S A, TORRES A G, et al. The aerobactin iron transport system genes in Shigella flexner are present within a pathogenicity island[J]. Mol Microbiol, 1999, 33(1): 63-73.
[19] FERDOUS M, ZHOU K, MELLMANN A, et al. Is shiga toxin-negative Escherichia coli O157: H7 enteropathogenic or enterohaemorrhagic Escherichia coli? a comprehensive molecular analysis using whole genome sequencing[J]. J Clin Microbiol, 2015, 53(11): 3530-3538.
[20] HARRIS S R, FEIL E J, HOLDEN M T, et al. Evolution of MRSA during hospital transmission and intercontinental spread[J]. Science, 2010, 327(5964): 469-474.
[21] KÖSER C U, HOLDEN M T, ELLINGTON M J, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak[J]. N Engl J Med, 2012, 366(24): 2267-2275.
(本文编辑:吴彬)
Data analysis of virulence factors in uropathogemic Escherichia coli NB8 by whole genome sequencing
WENG Xingbei1, MI Zuhuang2, WANG Chunxin3, ZHU Jianming4. 1.Department of Medical Laboratory, Ningbo First Hospital, Ningbo, 315010; 2.Department of Bioinformation, W uxi Clon-Gen Techonology Institute, Wuxi, 214026; 3.Department of Medical Laboratory, Wuxi People’s Hospital Aff liated to Nanjing Medical University, Wuxi, 214023; 4.Department of Medical Laboratory, Hangzhou Yuhang Hospital of Traditional Chinese Medicine, Hangzhou, 311106
Objective: To investigate virulence factors and pathogenic mechanisms in uropathogemic Escherichia coli (UPEC) NB8. Methods: UPEC NB8 was collected from urine sample of an inpatient in Ningbo First Hospital, in April, 2012. Sequencing gyrA and parC, BLASTn algorithm were performed to identify E.coli. Then, whole genome were sequenced by Illumina HiSeq and Ion Torrent PGM, and PCR was performed to fi ll gaps. Data mining of virulence factors were performed, and comparative genomics of virulence factors in NB8 and 9 strains of UPEC after whole genome sequencing were performed. Results: By whole genome sequencing, a putative chromosome sequence (14 gaps inside) was 4 550 369 bp, a putative plasmid sequence (33 gaps inside) was 635 377 bp. Some classes of virulence factors were positive in NB8, including adhesion (adhesin, type P fi mbriae, type I fi mbriae, Escherichia coli common pilus), iron uptake(aerobactin, heme, enterobactin), protease (secreted autotransporter toxin), toxin (hemolysin), Ag43, SHI-2 pathogenicity island protein. Conclusion: Virulence factors in UPEC NB8 played an important role in adhesion, invasion, cell viability, cell toxicity, and contribute to enhance the survival and virulence of the bacteria, so lead to urinary tract infection. Whole genome sequencing technology has the advantages of high speed, high throughput, low cost and high effi ciency, which is worth of promoting.
Escherichia coli; whole genome; multi-drug resistance; uropathogemic; virulence factor
R378.2
A
10.3969/j.issn.2095-9400.2016.11.004
2016-01-27
宁波市自然科学基金资助项目(2012A610186);浙江省中医药资助项目(2011ZB126)。
翁幸鐾(1980-),男,浙江宁波人,副主任技师。
猜你喜欢
现代畜牧兽医(2022年4期)2022-06-09
现代畜牧科技(2021年9期)2021-10-13
农药科学与管理(2019年9期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
中华灾害救援医学(2015年7期)2016-01-07
现代检验医学杂志(2015年2期)2015-02-06
现代检验医学杂志(2014年1期)2014-02-06
现代检验医学杂志(2014年5期)2014-02-02
现代检验医学杂志(2014年4期)2014-02-02
中国预防兽医学报(2013年1期)2013-09-11
