
圈养大熊猫粪便中微生物多样性的研究
2016-12-09 05:13王立志徐谊英
四川动物 2016年1期
王立志, 徐谊英
(四川农业大学动物营养研究所,动物抗病营养教育部重点实验室,四川雅安625014)
圈养大熊猫粪便中微生物多样性的研究
王立志*, 徐谊英
(四川农业大学动物营养研究所,动物抗病营养教育部重点实验室,四川雅安625014)
本试验采集了5只圈养成年大熊猫的新鲜粪便,用高通量测序技术,研究了大熊猫粪便中细菌和古菌的结构和组成。试验结果表明:圈养成年大熊猫粪便细菌主要由变形菌门Proteobacteria(74.45%)、厚壁菌门Firmicutes(15.66%)、拟杆菌门Bacteroidetes(4.34%)、蓝藻门Cyanophyta/色球藻纲Chroococcophyceae(4.01%)等组成,其中变形菌门主要以埃希氏杆菌属Esherichia/志贺氏菌属Shigella(49.84%)为主;厚壁菌门主要以梭菌属Clostridium(4.65%)为主;拟杆菌门主要以稳杆菌属Empedobacter(3.51%)为主;蓝藻门全部为未分类的色球藻纲(4.01%)。古菌主要由泉古菌门Crenarchaeota(55.99%)和广古菌门Euryarchaeota(42.33%)组成,其中优势古菌是热变形菌纲Thermoprotei(55.99%)中未分类的属和产甲烷菌属Methanogenium(24.70%)。
大熊猫;细菌;古菌;高通量测序
大熊猫Ailuropodamelanoleuca是我国特有的珍稀濒危动物,属食肉目,其消化器官和消化酶至今仍保留着肉食性动物的特点,但它已高度特化为以竹类植物性食物为生的动物。与其他动物一样,大熊猫胃肠道内定居着种群庞大的微生物,主要由细菌、真菌、原虫等构成(费立松等,2005;周杰珑等,2012),与其消化生理、健康、疾病的发生和发展有着密切的关系。大熊猫经常发生肠道疾病,尤其在圈养的情况下,肠道疾病是影响大熊猫健康的重要因素,这与大熊猫肠道菌群紊乱可能存在重要关系。但有关大熊猫肠道菌群的研究报道较少。
以往通过传统培养的方法和小规模测序技术,大熊猫肠道大量的细菌、少量的真菌和纤毛虫已被分离鉴定。而有关大熊猫粪便中古菌的研究还未见报道。本实验采用高通量测序技术对圈养成年大熊猫肠道细菌和古菌多样性进行分析,旨在了解成年大熊猫肠道正常细菌和古菌的多样性及其优势菌群,一方面有助于深入了解大熊猫的消化生理,为大熊猫肠道疾病的预防和治疗、大熊猫放归等提供科学的试验参考;另一方面有助于人们更加科学全面地认识动物胃肠道古菌的多样性,为研究动物源甲烷减排技术提供参考。
1 材料和方法
1.1 样品采集
在中国保护大熊猫研究中心雅安碧峰峡基地采集5只圈养成年雌性大熊猫(1只11岁,3只8岁,1只7岁)的新鲜粪便。所有采样的大熊猫均以竹子为主食,每天饲喂1.6 kg窝窝头,并饲喂少量的苹果、胡萝卜等,在采样之前已稳定采食该日粮30 d以上。清晨待大熊猫排粪后,立即无菌收集粪便中未污染的100 g左右的样品,-80 ℃保存备用。
1.2 粪便微生物总DNA提取
称取200 mg粪便样本至2 mL离心管中,向样本中加入1.4 mL粪便样品裂解缓冲液(ASL),混合均匀,用TIANGEN公司粪便DNA提取试剂盒(TIANamp Stool DNA Kit)抽提样品微生物总DNA。具体步骤参照试剂盒说明书进行。
1.3 PCR扩增
针对样品中细菌的16S rRNA V3~V4区,选用细菌特异性引物对515F(5’-GTGCCAGCMGCCGCGGTAA-3’)和806R(5’-GGACTACVSGGGTATCTAAT-3’)进行扩增;针对样品中古菌的16S rRNA V4~V5区,选用古菌特异性引物对A349F:(5’-GYGCASCAGKCGMGAAW-3’)和A806R:(5’-GGACTACVSGGGTATCTAAT-3’)进行扩增,扩增体系和扩增条件参照文献进行(Hristovetal.,2012)。
每个样品做3个重复,将同一样品的PCR产物混合后,用2%琼脂糖凝胶电泳检测,分别使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)、QIAquick Gel Extraction Kit凝胶回收试剂盒(Qiagen公司)切胶回收细菌和古菌PCR产物。细菌的5个扩增样品编号为C1(11岁)、C2(8岁)、C3(8岁)、C4(8岁)、C5(7岁);对应的古菌的5个扩增样品编号为P1(11岁)、P2(8岁)、P3(8岁)、P4(8岁)、P5(7岁)。
1.4 高通量测序
将细菌和古菌的PCR扩增产物送深圳千年盛世基因科技有限公司分别采用Illumina公司的MiSeq 250PE、MiSeq 300PE测序平台进行高通量测序。
1.5 高通量测序数据的处理及分析
用Mothur软件(Schlossetal.,2009)将测序所得原始数据去除引物和标签,并根据序列的重叠关系进行拼接;用QIIME软件(Caporasoetal.,2010)对拼接的序列进行过滤(去除序列平均碱基质量值小于Q25,未知碱基数大于6,序列长度小于200 bp和大于500 bp,引物信息与合成的引物碱基存在错配的序列);将以上过滤得到的序列与数据库(http://drive5.com/usearch/manual/uchime_algo.html)进行比对,去除其中的嵌合体,得到有效序列;使用UCLUST软件(Edgar,2010)对有效序列进行聚类,根据97%相似性水平(McKennaetal.,2008)将序列聚类成为聚类分类操作单元(operational taxonomic units, OTU),并挑选丰度最高的序列作为OTU代表序列;用PyNAST(Caporasoetal.,2010)将OTU代表序列与Greengenes 16S rRNA数据库(http://greengenes.lbl.gov/)进行比对,用RDP分类器(Coleetal.,2009)对OTU代表序列从门到属进行物种注释,然后绘制稀释曲线;最后,对所有样品的共享属进行分析,并用R软件(http://cran.rstudio.com)绘制共享热图。根据各个样品OTU的种类及其丰度,计算样品间的Unweighted Unifrac距离,并进行β多样性分析。
2 试验结果
2.1 大熊猫粪便中细菌的组成
本试验共获得属于细菌的有效序列84 614条,平均每个样品16 922条,共获得848个OTU。根据取样数量与OTU数目构建的稀释曲线(图1)可以看出,当取样数达到5时,稀释曲线的上升趋势明显变缓,这意味着本次试验5个样品数基本可以满足大熊猫粪便细菌多样性研究的需要。序列通过物种注释可分为20门42纲64目123科242属。在门、纲、目、科、属水平上平均相对丰度大于4%的分别为:变形菌门(74.46%)、厚壁菌门(15.66%)、拟杆菌门(4.34%)和蓝藻门(4.01%)(图2:A);γ-变形菌纲(74.02%)、芽孢杆菌纲(8.79%)、梭菌纲(6.52%)、黄杆菌纲(4.13%)、色球藻纲(4.01%)(图2:B);肠杆菌目(67.04%)、芽孢杆菌目(7.44%)、梭菌目(6.52%)、假单胞菌目(5.97%)、黄杆菌目(4.13%)(图2:C);肠杆菌科(67.04%)、莫拉菌科(4.75%)、梭菌科(4.68%)、黄杆菌科(4.13%)、未分类的色球藻纲(4.01%)(图2:D);埃希氏杆菌属/志贺氏菌属(49.84%)、不动杆菌属(4.73%)、梭菌属(4.65%)、木霉(4.01%)(图2:E)。
2.2 大熊猫粪便中古菌的组成
本试验共获得属于古菌的有效序列5460条,平
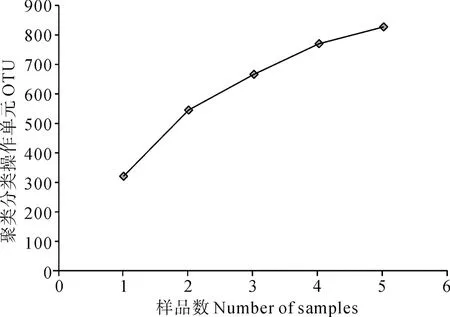
图1 基于采样数量的OTU稀疏分析
均每个样品1092条。序列经物种注释后得到295个OTU。根据取样数量与OTU数目构建的稀释曲线(图3)可以看出,5个样品数基本可以满足大熊猫粪便古菌多样性研究的需要。序列的物种注释结果显示,全部古菌OTU可分为3门8纲11目14科15属。在门水平上包括泉古菌门(55.99%)、广古菌门(42.33%)和未鉴定的古菌(1.68%)(表1)。泉古菌门主要由未分类的古菌组成,还包括Fervidicoccus和硫化叶菌属,但分别只占总序列数的0.03%和0.02%。广古菌门主要由甲烷短杆菌属(21.22%)、热裸单孢菌属(5.70%)、甲烷微菌属(2.48%)、甲烷球形菌属(0.76%)、甲烷微球菌属(0.21%)、甲烷粒菌属(0.04%)组成。
2.3 共享属分析
从属的水平对样品间共享的细菌种类和丰度进行了分析,结果发现,5只大熊猫之间的细菌共享属共有31个(图4)。从图中可以看出,样品C1中哈夫尼菌属等12个属、样品C2中鞘氨醇单胞菌属等12个属、样品C3中Luteibacter等8个属、样品C4中鞘氨醇单胞菌属等10个属、样品C5中假单胞菌属等12个属分别高于它们各自在5个样品中的平均含量(红色部分,颜色越深,高出平均值越多)。大熊猫肠道古菌的共享属只有4个(表2),即甲烷短杆菌属、甲烷微菌属、甲烷微球菌属和热裸单胞菌属,且前3个共享属均属于产甲烷菌。
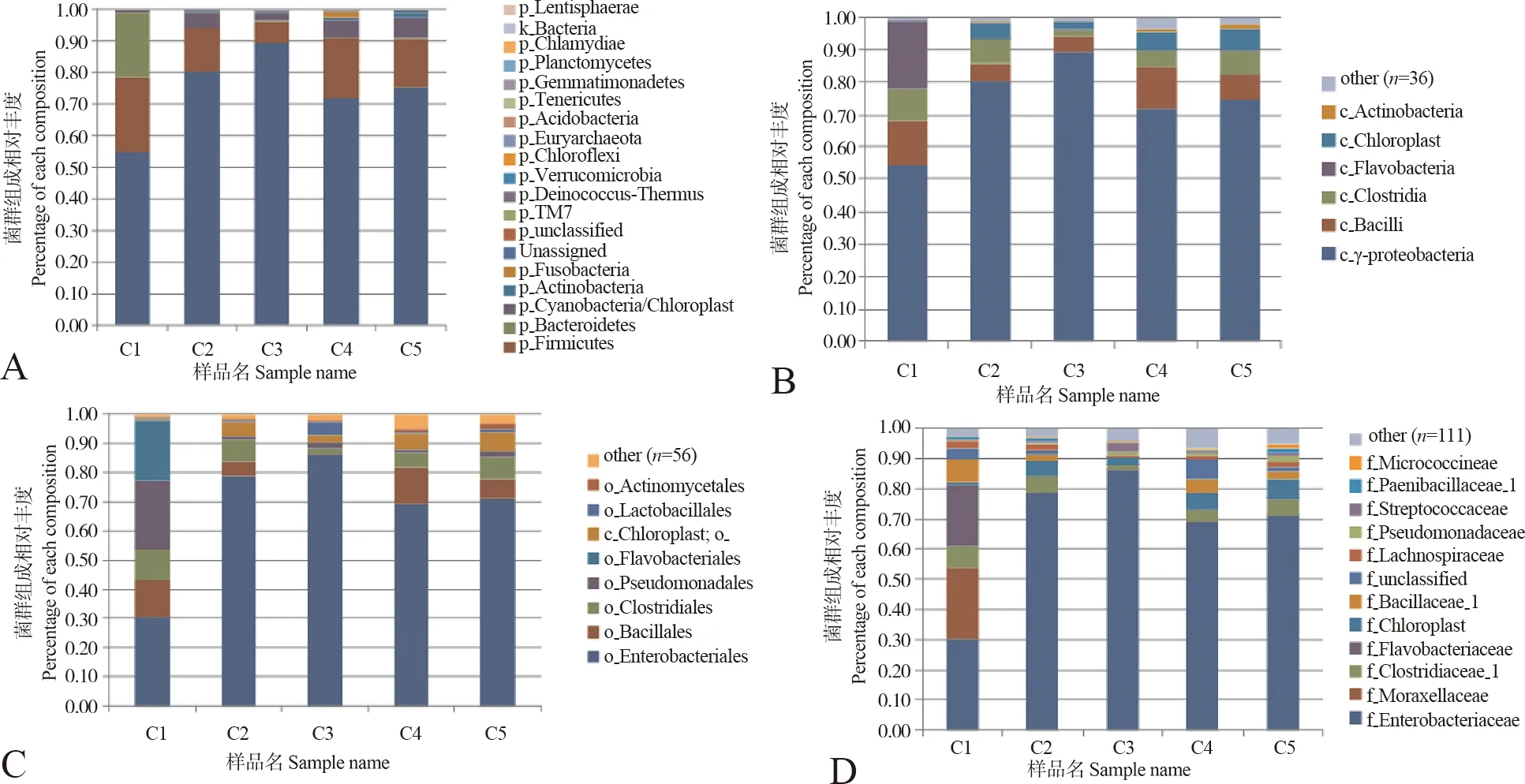
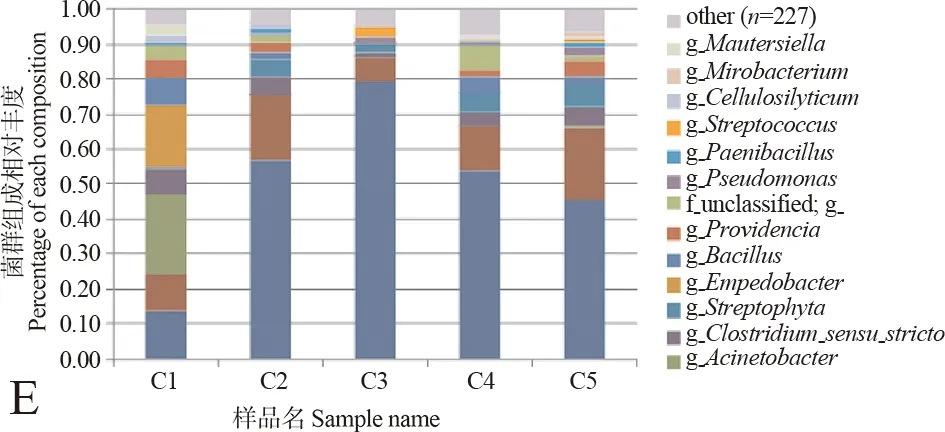
图2 基于各水平上的微生物物种组成
A. 基于门水平的物种组成, B. 基于纲水平的物种组成, C. 基于目水平的物种组成, D. 基于科水平的物种组成, E. 基于属水平的物种组成; 在不同分类水平上平均相对丰度低于1%的分类统一合并为“other”。
A, B, C, D and E presented the bacterial community composition at phylum, class, order, family and genus level, respectively; The taxa whose abundance were less than 1% at different taxonomic level were merged into “other”.
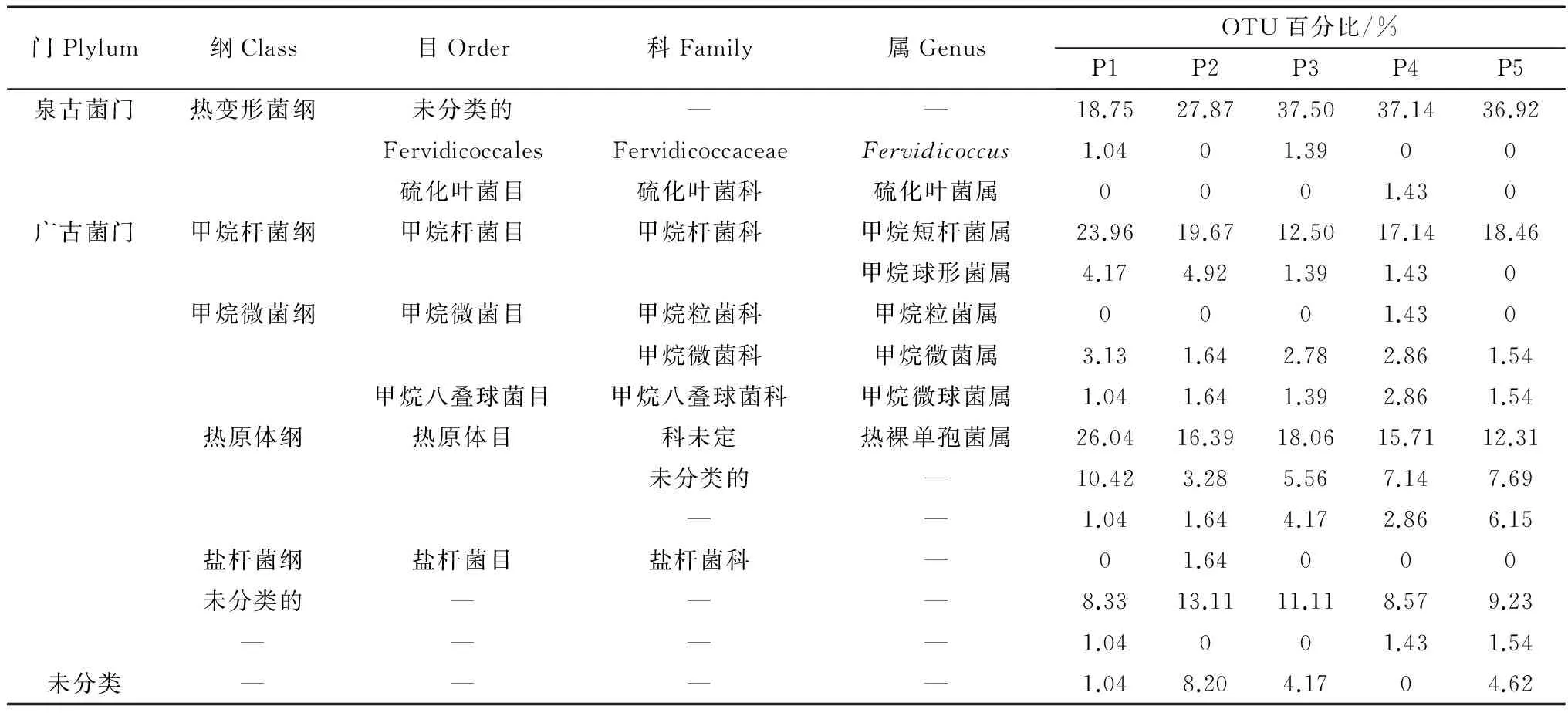
表1 圈养大熊猫粪便中古菌的组成
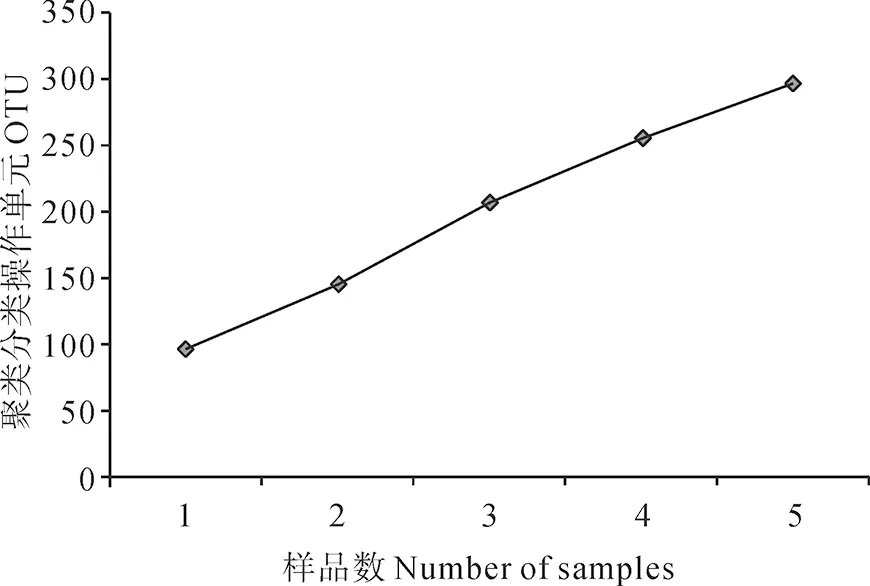
图3 基于采样数量的古菌OTU稀疏分析
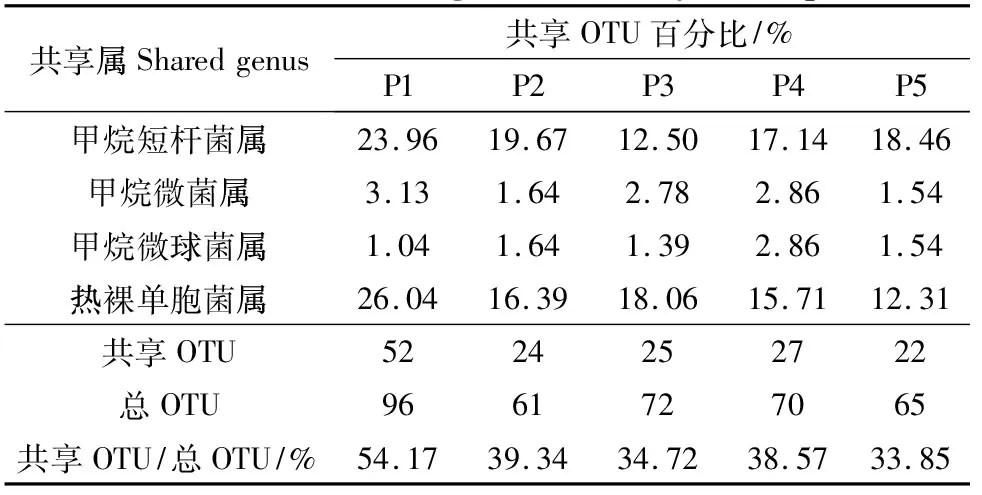
表2 样品间古菌共享属
2.4 β多样性分析
根据大熊猫粪便中细菌、古菌各自OTU的种类及丰度,采用 QIIME软件,计算得到了样品两两间的Unweighted Unifrac距离。根据细菌的种类和丰度计算出的大熊猫不同个体间Unweighted Unifrac距离最大为0.23(C1与C3之间),最小为0.04(C2与C5之间),平均值为0.10±0.08,说明大熊猫粪便中细菌的结构与组成相似度很高;基于Unweighted Unifrac距离矩阵,使用非加权组平均法(unweighted pair group method with arithmetic mean, UPGMA)对样品进行聚类分析(图5:A),除了1只老年个体(C1)与其他个体间聚集度较低外,其他样品聚集度都很高。根据古菌计算出的个体间Unweighted Unifrac距离最大为0.56(P1与P4之间),最小为0.20(P3与P5之间),平均值为0.36±0.12,说明大熊猫粪便中古菌的结构与组成相似度较细菌低,UPGMA聚类结果见图5B,老年个体(P1)与其他个体间聚集度较低。
3 讨论
以大熊猫粪便为研究对象,张志和等(1995)、熊焰等(2000)以传统的培养法,Wei等(2007)和何廷美等(2012)运用16S rDNA小规模测序技术都曾研究过大熊猫肠道菌群多样性。本实验采用高通量测序技术共获得了84 614条属于细菌的有效序列,经物种注释共得到848个OTU,鉴定出的细菌种类远多于以往的研究结果。
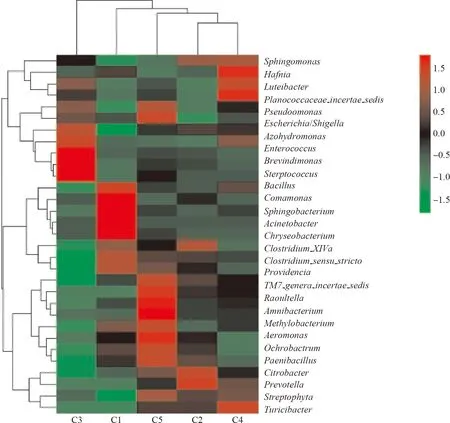
图4 样本间细菌共享属的聚类热图
Fig. 4 The clustering of bacterial genera shared by all samples
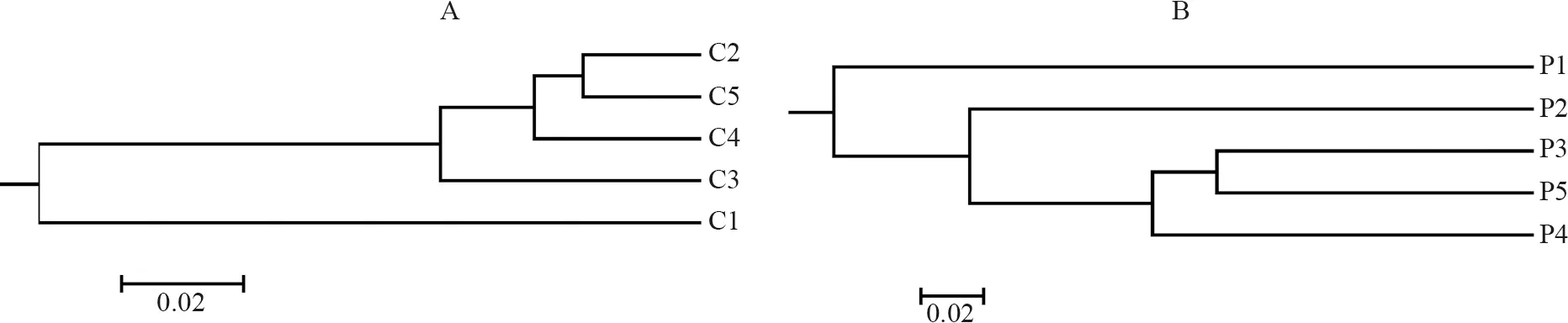
图5 大熊猫样品的UPGMA聚类图
本研究发现,在门水平上,大熊猫粪便中细菌以变形菌门为主,与Fang等(2012)的研究结果基本一致,但与其他动物有很大差异。一般食草动物(牛、梅花鹿)胃肠道微生物都以厚壁菌门和拟杆菌门为主(Jami & Mizrahi,2012;李志鹏,2013)。大熊猫虽然也以植物——竹子为主食,在其肠道内也检测到了纤维消化菌(Zhuetal.,2011),但它并不具备一个专门消化植物粗纤维的器官,其肠道也不适宜消化植物纤维的微生物大量定植繁衍,这也许就是大熊猫对竹子消化率非常低,肠道厚壁菌门和拟杆菌门微生物含量低的原因。然而,Zhu等(2011)和Tun等(2014)的研究结果都显示大熊猫粪便中的微生物以厚壁菌门为主。Zhu等(2011)的研究对象是8只圈养大熊猫和7只野生大熊猫,这15个样品的总体平均是以厚壁菌门为主,但是如果单看圈养的大熊猫,变形菌门是大熊猫粪便中的优势菌,与本研究的结果相同。Tun等(2014)研究发现,2只成年和2只老年大熊猫粪便中,丰度最高的是厚壁菌门(42%~79%),其次是变形菌门(21%~58%)。造成这种差异的原因可能主要在于DNA提取方法的差异。厚壁菌门的细菌DNA在提取时,需要在95 ℃处理10 min,同时需要机械破壁。本实验在提取DNA时没有采取这些措施,因此有可能导致厚壁菌门的丰度被低估。
本次试验在大熊猫粪便中发现大量蓝细菌,以往还未见这类细菌在其他动物胃肠道中被检测到的报道。蓝细菌为原核生物,是一种能产生氧气的光合细菌,主要分布于河流、湖沼和海洋等水体中(Esquenazietal.,2011)。四川气候潮湿,这就使得大量的蓝细菌可以滋生于竹子茎叶上,当其随竹子被大熊猫采食进入体内后,由于大熊猫对食物的消化率低,大量未被消化的蓝细菌就会随粪便排出体外。因此,虽然大熊猫粪便菌群中蓝细菌的相对丰度较高,也许它并不发挥与消化吸收有关的任何作用,而仅仅只是作为一种过路菌存在于大熊猫肠道中。这种推测是否正确,还有待进一步的研究确认。
本试验是首次分析大熊猫粪便中古菌多样性的研究报道。试验结果显示,大熊猫粪便中丰度最高的古菌是泉古菌门的热变形菌。这一结果与其他动物胃肠道古菌的组成有较显著的差异。目前的研究结果显示,动物胃肠道中的古菌绝大部分属于广古菌门的产甲烷菌,只有少量研究发现动物胃肠道中存在热变形菌。如Kim等(2011)在研究RDP数据库中来自全球各地瘤胃微生物的序列时发现,仅有11条序列属于热变形菌,比例非常低。本试验检测出如此多热变形菌的原因可能有两点:第一,动物胃肠道中原本就存在一定量的热变形菌,以往受研究技术的限制,未能将之检测出来;第二,大熊猫自身特殊的消化系统与饮食习惯相互作用,导致其消化道内微生物(特别是古菌)有别于其他动物。这种推测是否正确还有待进一步的研究。
本试验共检测出5个属的产甲烷菌,其中大部分的产甲烷菌属于甲烷短杆菌属,这与其他动物胃肠道内产甲烷菌的优势菌群基本类似。研究发现,加拿大安大略地区及爱德华王子岛以玉米为主要饲料的奶牛瘤胃内优势的产甲烷菌是甲烷短杆菌属(51.97%、50%),南美地区麝雉嗉囊中、挪威驯鹿瘤胃内、泌乳期的荷斯坦奶牛及娟珊牛瘤胃内、澳大利亚袋鼠前肠(春季)中甲烷短杆菌属的数量也相当丰富(Wrightetal.,2007,2009;Evansetal.,2009;Sundsetetal.,2009a,2009b;Kingetal.,2011)。但也有与本实验结果不一致的报道,Fouts等(2012)发现甲烷短杆菌属和甲烷球形菌属是牛瘤胃内的优势产甲烷菌,但本试验的样品P5中并没有检测出甲烷球形菌属,并且其他4个样品中甲烷球形菌属的含量也较少;Min等(2014)研究肉山羊瘤胃内产甲烷菌,结果发现甲烷短杆菌属、甲烷球形菌属、Methanobacteriaceae是其瘤胃中的优势产甲烷菌;Huang等(2012)研究显示,中国青藏高原奶牛和牦牛瘤胃内优势菌群是Methanomassiliicoccus;Chaudhary等(2012)研究发现,印度地区饲喂小麦秸秆的莫拉水牛瘤胃主要的产甲烷菌是甲烷粒菌属。到目前为止,影响动物消化道产甲烷菌多样性的主要原因仍然不清楚,但是许多因素常常单独地或者综合地影响着产甲烷菌的组成和数量,如宿主、日粮和地理环境等。因此,本试验的结果与其他动物消化道内产甲烷菌多样性有一定的差异,这可能是大熊猫与消化道产甲烷菌共同选择的结果,也可能是大熊猫的生存环境影响了其消化道产甲烷菌的多样性,具体原因还不清楚。
本试验中,不论是细菌还是古菌的β多样性分析都显示,11岁的大熊猫个体与其他4只7~8岁的大熊猫个体之间,粪便微生物的结构和组成上差异性较大。Tun等(2014)的研究也表明,Actinobacteria门的某些组分仅出现在成年大熊猫粪便中,在老年大熊猫粪便中检测不到,且老年大熊猫的微生物多样性指数较成年个体的低。这是否说明年龄对大熊猫胃肠道微生物的多样性存在显著影响,还需要进一步研究。因为所有有关大熊猫胃肠道微生物的研究样本数都很少,今后的研究应特别注意这一点。
费立松, 杨光友, 张志和, 等. 2005. 大熊猫胃内纤毛虫检测初报[J]. 动物学报, 51(3): 526-529.
何廷美, 崔婷婷, 钟志军, 等. 2012. 成年大熊猫肠道菌群多样性的16S rDNA-RELP分析[J]. 中国兽医科学, 42(11): 1121-1127.
李志鹏. 2013. 梅花鹿瘤胃微生物多样性与优势菌群分析[D]. 北京: 中国农业科学院.
熊焰, 李德生, 王印, 等. 2000. 卧龙自然保护区大熊猫粪样菌群的分离鉴定与分布研究[J]. 畜牧兽医学报, 31(2): 165-170.
张志和, 何光昕, 张安居, 等. 1995. 大熊猫肠道正常菌群的研究[J]. 兽类学报, 15(3): 170-175.
周杰珑, 刘丽, 王家晶, 等. 2012. 春季动物园大熊猫粪便可培养真菌鉴定与分析[J]. 西南林业大学学报, 32(1): 75-78.
Caporaso JG, Kuczynski J, Stombaugh J,etal. 2010. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 7(5): 335-336.
Chaudhary PP, Sirohi SK, Saxena J. 2012. Diversity analysis of methanogens in rumen ofBubalusbubalisby 16S riboprinting and sequence analysis[J]. Gene, 493(1): 13-17.
Cole JR, Wang Q, Cardenas E,etal. 2009. The ribosomal database project: improved alignments and new tools for rRNA analysis[J]. Nucleic Acids Research, 37: 141-145.
Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics, 26(19): 2460-2461.
Esquenazi E, Jones AC, Byrum T,etal. 2011. Temporal dynamics of natural product biosynthesis in marine cyanobacteria[J]. Proceedings of the National Academy of Sciences, 108(13): 5226-5231.
Evans PN, Hinds LA, Sly LI,etal. 2009. Community composition and density of methanogens in the foregut of the tammar wallaby (Macropuseugenii)[J]. Applied and Environmental Microbiology, 75(8): 2598-2602.
Fang W, Fang ZM, Zhou P,etal. 2012. Evidence for lignin oxidation by the giant panda fecal microbiome[J]. PLoS ONE, 7(11): e50312.
Fouts DE, Szpakowski S, Purushe J,etal. 2012. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen[J]. PLoS ONE, 7(11): e48289.
Hristov AN, Callaway TR, Lee C,etal. 2012. Rumen bacterial, archaeal, and fungal diversity of dairy cows in response to ingestion of lauric or myristic acid[J]. American Society of Animal Science, 90(12): 4449-4457.
Huang XD, Tan HY, Long RJ,etal. 2012. Comparison of methanogen diversity of yak (Bosgrunniens) and cattle (Bostaurus) from the Qinghai-Tibetan plateau, China[J]. BMC Microbiology, 12: 237.
Jami E, Mizrahi I. 2012. Composition and similarity of bovine rumen microbiota across individual animals[J]. PLoS ONE, 7(3): e33306.
Kim M, Morrison M, Yu ZT. 2011. Status of the phylogenetic diversity census of ruminal microbiomes[J]. FEMS Microbiology Ecology, 76(1): 49-63.
King EE, Smith RP, St-Pierre B,etal. 2011 Differences in the rumen methanogen populations of lactating Jersey and Holstein dairy cows under the same diet regimen[J]. Applied and Environmental Microbiology, 77(16): 5682-5687.
McKenna P, Hoffmann C, Minkah N,etal. 2008. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis[J]. PLoS Pathogens, 4(2): e20.
Min BR, Solaiman S, Shange R,etal. 2014. Gastrointestinal bacterial and methanogenic archaea diversity dynamics associated with condensed tannin-containing pine bark diet in goat using 16S rDNA amplicon pyrosequencing[J/OL]. International Journal of Microbiology: 1-11[2015-04-19]. http://www.hindawi.com/journals/ijmicro/2014/141909.DOI: 10. 1155/2014/141909.
Schloss PD, Westcott SL, Ryabin T,etal. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities[J]. Applied Environmental Microbiology, 75(23): 7537-7541.
Sundset MA, Edwards JE, Cheng YF,etal. 2009a. Molecular diversity of the rumen microbiome of norwegian reindeer on natural summer pasture[J]. Microbial Ecology, 57(2): 335-348.
Sundset MA, Edwards JE, Cheng YF,etal. 2009b. Rumen microbial diversity in Svalbard reindeer, with particular emphasis on methanogenic archaea[J]. FEMS Microbiology Ecology, 70(3): 553-562.
Tun HM, Mauroo NF, Yuen CS,etal. 2014. Microbial diversity and evidence of novel homoacetogens in the gut of both geriatric and adult giant pandas (Ailuropodamelanoleuca)[J]. PLos ONE, 9(1): e79902.
Wei G, Lu H, Zhou Z,etal. 2007. The microbial community in the feces of the giant panda (Ailuropodamelanoleuca) as determined by PCR-TGGE profiling and clone library analysis[J]. Microbial Ecology, 54: 194-202.
Wright AG, Auckland CH, Lynn DH. 2007. Molecular diversity of methanogens in feedlot cattle from Ontario and Prince Edward Island, Canada[J]. Applied and Environmental Microbiology, 73(13): 4206-4210.
Wright AG, Northwood KS, Obispo NE. 2009. Rumen-like methanogens identified from the crop of the folivorous south American bird, the hoatzin (Opisthocomushoazin)[J]. ISME Journal, 3(10): 1120-1126.
Zhu LF, Wu Q, Dai JY,etal. 2011. Evidence of cellulose metabolism by the giant panda gut microbiome[J]. PNAS, 108(43): 17714-17719.
Diversity of Microorganism in the Feces of Captive Giant Pandas
WANG Lizhi*, XU Yiying
(Animal Nutrition Institute of Sichuan Agricultural University, Key Laboratory for Animal Disease-Resistance Nutrition of Ministry of Education, Ya’an, Sichuan Province 625014, China)
The structures and compositions of bacteria and archaeal in the feces of five adult captive giant pandas were studied using high-throughput sequencing technology. The results showed that the bacteria mainly consisted of Proteobacteria (74.45% of total sequences), Firmicutes (15.66%), Bacteroidetes (4.34%), and Cyanophyta/Chroococcophyceae (4.01%) at the phylum level. Within the phylum Proteobacteria,Escherichia/Shigella(49.84%) was the most abundant genus. Within the phylum Firmicutes,Clostridium(4.65%) was the most abundant genus. Within the phylum Bacteroidetes,Empedobacter(3.51%) was the most abundant genus. Within the phylum Cyanophyta//Chroococcophyceae, only the unclassified Chroococcophyceae (4.01%) was detected. The archaea mainly consisted of Crenarchaeota (55.99%) and Euryarchaeota (42.33%) at the phylum level, and the unclassified Thermoprotei (55.99%) andMethanogenium(24.70%) were the two dominant memberships at the genus level.
giant panda; bacterium; archaea; high-throughput sequencing
10.11984/j.issn.1000-7083.20150140
2015-04-19 接受日期:2015-06-23
四川省科技厅国际合作项目(2013HH0043畜禽温室气体排放量检测及减排技术研究)
王立志(1974—), 男, 博士, 副教授, 硕士生导师, 主要从事动物营养学研究
*通信作者Corresponding author, E-mail:wanglizhi@aliyun.com
Q959.8
A
1000-7083(2016)01-0017-07
猜你喜欢
中国感染控制杂志(2022年11期)2022-11-24
土壤学报(2022年3期)2022-08-26
大自然探索(2022年5期)2022-07-11
知识就是力量(2022年6期)2022-06-16
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
智富时代(2019年5期)2019-07-05
智富时代(2019年5期)2019-07-05
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
