
微波促进水杨醛肟一锅法制备2-二氟甲氧基苯腈
2016-12-02 03:03华明清刘威旱
高等学校化学学报 2016年3期
王 韬, 华明清, 刘威旱, 黄 燕, 张 岐,2
(1. 江苏大学化学化工学院, 镇江 212013; 2. 海南大学海南省精细化工重点实验室, 海口 570228)
微波促进水杨醛肟一锅法制备2-二氟甲氧基苯腈
王 韬1, 华明清1, 刘威旱1, 黄 燕1, 张 岐1,2
(1. 江苏大学化学化工学院, 镇江 212013; 2. 海南大学海南省精细化工重点实验室, 海口 570228)
以氯二氟乙酸钠为二氟甲基化试剂, 碳酸钾为碱, 实现了微波促进水杨醛肟一锅脱水成腈及二氟甲基化反应, 以中等收率获得了8个2-二氟甲氧基苯腈类化合物, 其中7个为新化合物. 利用核磁共振波谱、 红外光谱和高分辨质谱等手段对目标产物进行了表征. 讨论了二氟甲基化试剂、 碱和溶剂的种类、 微波功率、 反应温度和时间对反应的影响. 确定了最优反应条件: 含有不同取代基的水杨醛肟、 氯二氟乙酸钠、 碳酸钾和N,N-二甲基甲酰胺的摩尔比为1∶1.5∶1.5∶13, 微波功率300 W, 反应温度85 ℃, 反应时间20 min. 结合对比实验, 提出了可能的反应机理.
水杨醛肟; 2-二氟甲氧基苯腈; 二氟甲基化; 微波
氟原子具有电负性高、 极化率低和体积相对较小等特性, 因而含氟化合物具有独特的物理、 化学及生理性能, 在医药、 农药和材料领域得到广泛应用[1]. 在含氟基团中, 二氟甲基(—CF2H)由于具有亲脂性、 膜通透性、 水溶性及代谢稳定性等优良的生理活性而备受关注[2~17]. 用二氟甲基化试剂对化合物进行二氟甲基化反应可以制得含二氟甲基化合物.
芳腈是一类重要的化工原料和有机合成中间体, 存在于多种医药、 染料、 农药和天然产物中, 并能通过一系列官能团转化生成酰胺类及杂环类化合物等[18~20]. 芳腈类化合物的合成方法很多[21~26], 经典的方法为Rosenmund-von Braun取代法, 即卤代芳香烃与氰化亚铜进行置换反应得到芳腈. 由于Rosenmund-von Braun取代法需在高温条件下使用高毒性的氰化亚铜, 限制了其在有机合成中的应用. Hell等[27]、 李小六等[28]和邝代治等[29]报道了芳香醛肟经金属或非金属催化剂催化脱水生成芳腈的反应, 在微波辅助下可快速, 高效制备芳腈类化合物. 通常, 常规方法合成2-二氟甲氧基苯腈及其衍生物需要使用成本较高的2-羟基苯腈与三甲基溴二氟甲基硅烷[30].
本文以水杨醛肟为底物, 以氯二氟乙酸钠为二氟甲基化试剂, 碳酸钾为碱, 在微波促进下, 经一锅多步反应制备了2-二氟甲氧基苯腈及其衍生物. 利用核磁共振波谱、 红外光谱和高分辨质谱等手段对目标产物进行了表征; 并讨论了二氟甲基化试剂、 碱、 溶剂、 微波功率、 反应温度和时间的影响.
1 实验部分
1.1 试剂与仪器
水杨醛(1a)、 2-羟基苯乙酮、 盐酸羟胺、 碳酸氢钠、 碳酸钾、 甲苯(PhMe)、 乙腈(MeCN)、 乙酸乙酯和石油醚均为分析纯, 国药集团化学试剂有限公司; 5-氯水杨醛(1b)、 5-溴水杨醛(1c)和4-二乙氨基水杨醛(1d)的纯度为99%, 萨恩化学技术(上海)有限公司; 5-甲基水杨醛(1e)、 4-甲氧基水杨醛(1f)和3-甲基水杨醛(1h)的纯度为98%, 东京化成工业株式会社; 5-硝基水杨醛和氯二氟乙酸钠的纯度均为98%, 北京偶合科技有限公司; N,N-二甲基甲酰胺(DMF)和无水乙醇(EA)均为分析纯, 成都市科龙化工试剂厂; N,N-二甲基乙酰胺(DMAc)和N-甲基吡咯烷酮(NMP)均为化学纯, 上海市晶纯试剂有限公司; 柱层析硅胶(精制型), 青岛海洋化工厂分厂.
XH100B型微波催化合成/萃取仪, 北京祥鹄科技有限公司; X-5型控温型显微熔点测定仪, 北京泰克仪器有限公司; Nicolet Nexus 470型傅里叶变换红外光谱仪(FTIR), 美国Nicolet公司; AVANCE Ⅱ 400 MHz型核磁共振波谱仪(NMR), 瑞士Bruker公司; maXis型超高分辨飞行时间质谱仪(HRMS), 德国Bruker公司.
1.2 2-二氟甲氧基苯腈的制备
目标化合物3a~3h的合成路线见Scheme 1.
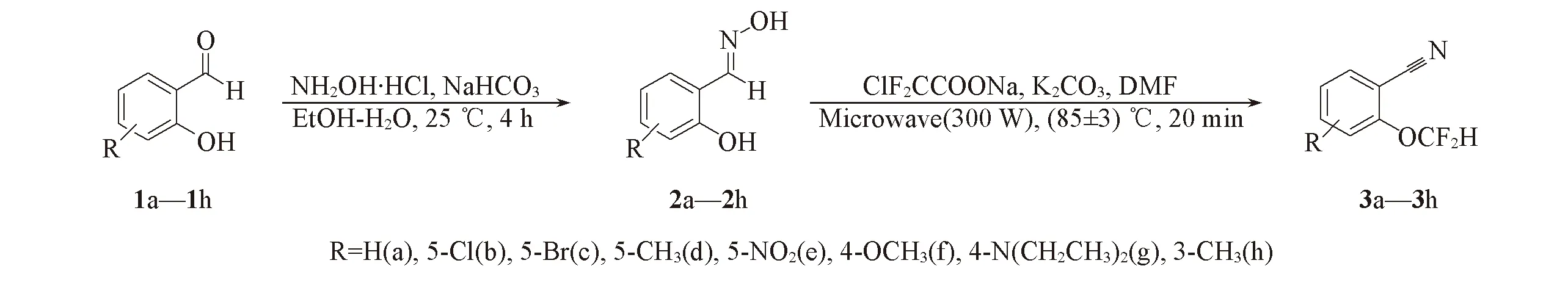
Scheme 1 Synthetic route of the target compounds
向50 mL三口烧瓶中加入化合物2a~2h(2 mmol)、 碳酸钾(3 mmol, 过量50%)、 氯二氟乙酸钠(3 mmol, 过量50%)和N,N-二甲基甲酰胺(DMF, 2 mL), 搅拌均匀后置于微波催化合成仪中, 设定功率为300 W, 温度为85 ℃, 反应20 min; 冷却后, 向反应体系中分别加入10 mL乙酸乙酯和10 mL水, 将有机相分离并用10 mL水洗3次. 有机相经无水硫酸镁干燥, 过滤, 真空除去溶剂, 粗产品经硅胶柱层析纯化[洗脱剂: V(乙酸乙酯)∶V(石油醚)=1∶40], 得到产物2-二氟甲氧基苯腈(3a)、 5-氯-2-二氟甲氧基苯腈(3b)、 5-溴-2-二氟甲氧基苯腈(3c)、 5-甲基-2-二氟甲氧基苯腈(3d)、 5-硝基-2-二氟甲氧基苯腈(3e)、 4-甲氧基-2-二氟甲氧基苯腈(3f)、 4-二乙氨基-2-二氟甲氧基苯腈(3g)和3-甲基-2-二氟甲氧基苯腈(3h). 底物为2a时, 分离出少量2-羟基苯腈(4a), 白色固体, m.p. 96~99 ℃(文献值[37]: 96~98 ℃). 产物经1H NMR,13C NMR,19F NMR, IR和HRMS确定结构. 化合物3a~3h的理化性质见表1, 核磁共振表征结果见表2.

Table 1 Appearance, yields, melting points, IR and HRMS data of compounds 3a—3h
Continued
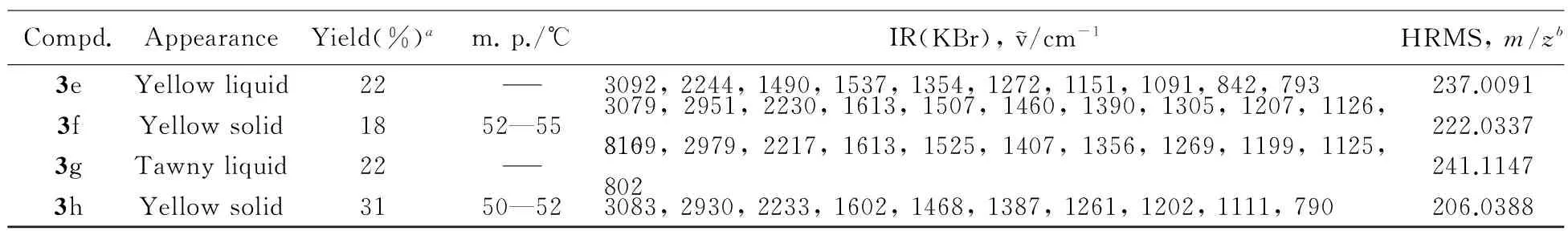
Compd.AppearanceYield(%)am.p./℃IR(KBr),v/cm-1HRMS,m/zb3eYellowliquid223092,2244,1490,1537,1354,1272,1151,1091,842,793237.00913fYellowsolid1852—553079,2951,2230,1613,1507,1460,1390,1305,1207,1126,816222.03373gTawnyliquid223109,2979,2217,1613,1525,1407,1356,1269,1199,1125,802241.11473hYellowsolid3150—523083,2930,2233,1602,1468,1387,1261,1202,1111,790206.0388
a. Isolated yield; b. HRMS for compound 3f is [M+H]+, for others are [M+Na]+.

Table 2 1H NMR, 13C NMR and 19F NMR data for compounds 3a—3h*
* NMR solvent for compound 3f is DMSO-d6, for others are CDCl3.
2 结果与讨论
2.1 反应条件的优化
2.1.1 反应温度、 时间及微波功率的影响 以水杨醛肟2a为原料, 氯二氟乙酸钠为二氟甲基化试剂, DMF为溶剂, 考察了不同反应温度、 反应时间及微波功率对该反应的影响, 结果见表3. 由表3可知, 最佳反应温度为85 ℃, 温度过低和过高都不利于化合物3a的生成, 当温度达到100 ℃时, 副产物2-羟基苯腈(4a)增多(表3, Entries 1~5).
在最佳反应温度下, 反应时间对于产物3a收率的影响较显著. 当反应时间在20 min之内, 化合物3a的收率随着时间的增加而缓慢提高, 在20 min时达到最高的50%收率(表3, Enteries 3, 6和7); 而当反应时间超过20 min时, 化合物3a的收率随着时间的增加而迅速降低(表3, Enteries 8和9).
微波功率对于该反应也有较大影响, 在加热条件(非微波环境)或微波功率为200 W时, 该反应未生成产物(表3, Entries 10和11); 当微波功率为300 W时, 以50%收率获得产物3a(表3, Entry 3); 当微波功率为400 W时, 产物3a的收率降至46%(表3, Entry 12). 由此确定最优微波功率为300 W.

Table 3 Effects of heating power, temperature and time on the reactiona
a. Unless otherwise noted, all the reactions were carried out with compound 2a(2 mmol), ClF2CCOONa(3 mmol), K2CO3(3 mmol), 2 mL DMF; b. isolated yield.
2.1.2 溶剂、 碱及二氟甲基化试剂对反应的影响 在最佳反应温度、 时间及微波功率下, 考察了不同溶剂对该反应的影响, 结果见表4.
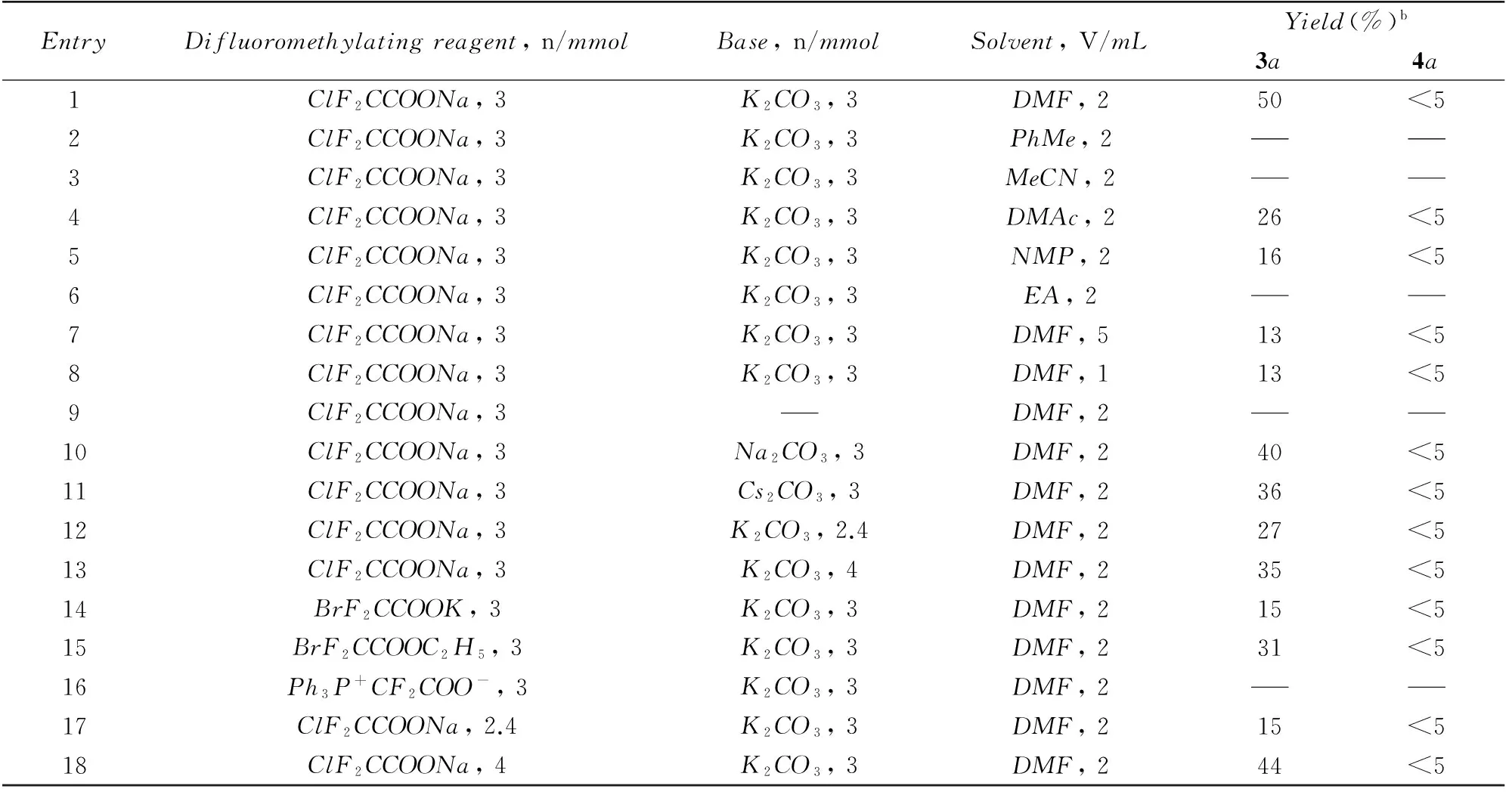
Table 4 Optimization of reaction conditionsa
a. Unless otherwise noted, all the reactions were carried out with compound 2a(2 mmol); b. isolated yield.
由表4可知, 溶剂对产物3a的生成影响较大. 在甲苯(PhMe)、 乙腈(MeCN)和乙醇(EA)中, 无产物生成(表4, Entries 2, 3和6); 在N,N-二甲基乙酰胺(DMAc)和N-甲基吡咯烷酮(NMP)中, 产物3a收率偏低, 仅为26%和16%(表4, Entries 4和5); 在DMF中产物3a的收率为50%(表4, Entry 1). 同时, 反应物2a的浓度变化也影响反应结果(表4, Entries 7和8). 实验结果表明, 反应的最优溶剂为DMF, 反应物2a的最优浓度为1 mol/L.
考察了不同碱对该反应的影响. 由表4可知, 不加入碱时反应不能进行. 对不同碳酸盐的筛选结果显示, 当使用碳酸钠或碳酸铯代替碳酸钾时, 产物3a的收率均有所降低(表4, Entries 9~11); 而碱的使用量也影响反应效果(表4, Entries 12和13). 基于实验结果选定最优碱为碳酸钾, 其用量为理论量过量50%.
以溴二氟乙酸钾(BrF2CCOOK)和溴二氟乙酸乙酯(BrF2CCOOC2H5)分别作为二氟甲基化试剂时, 反应能够顺利进行, 但产物3a的收率较低(表4, Entries 14和15); 而以三苯基膦二氟乙酸盐(Ph3P+CF2COO-)为二氟甲基化试剂时, 则无产物生成(表4, Entry 16). 同时, 二氟甲基化试剂的用量也影响产物3a的生成(表4, Entries 17和18). 实验选择最优的二氟甲基化试剂及用量为氯二氟乙酸钠过量50%.
综上, 确定了最优反应条件: 含有不同取代基水杨醛肟、 氯二氟乙酸钠、 碳酸钾和N,N-二甲基甲酰胺的摩尔比为1∶1.5∶1.5∶13, 设定微波功率为300 W, 温度为85 ℃, 反应20 min.
2.2 反应的适用性
在最优反应条件下, 研究了该反应体系的底物普适性, 结果见表1. 首先, 研究了以5位含不同取代基的水杨醛肟(化合物2b, 2c, 2d和2e)作为底物时的产率. 由表2可知, 5位上的卤素取代基(氯或溴)对于该反应无太大影响, 产物3b和3c的收率与3a相差不大; 当5位上含有供电子的甲基时, 产物3d的收率降至33%; 当5位上含有吸电子的硝基时, 产物3e的收率降至22%; 当3位, 4位含有供电子取代基时, 产物3f, 3g及3h的收率都明显降低.
在相同条件下, 以2-羟基苯乙酮肟为底物时, 无二氟甲基化产物生成, 仅以76%的收率得到脱水成环产物3-甲基-1,2-苯并异噁唑浅黄色液体, MS(C8H7NO+H+计算值), m/z: 134.2(134.1)[38].
将水杨醛(1 mmol)、 盐酸羟胺(1.5 mmol)和氯二氟乙酸钠(1.5 mmol)混合、 加入K2CO3(3 mmol)和2 mL(25.9 mmol) DMF, 在85 ℃和微波功率300 W条件下, 用一锅微波法反应40 min, 仅以24%的收率获得水杨醛肟白色固体, m. p. 57~59 ℃(文献值[31]: 56~57 ℃).
2.3 机理探讨
为深入研究微波促进水杨醛肟一锅脱水成腈及二氟甲基化制备2-二氟甲氧基苯腈反应的历程, 尝试不加入氯二氟乙酸钠进行反应, 结果显示, 水杨醛肟和2-羟基苯乙酮肟都未发生反应. 此结果证实氯二氟乙酸钠参与了醛肟转变为腈及2-羟基苯乙酮肟转变为3-甲基-1,2-苯并异噁唑的过程. 还尝试以2-羟基苯腈4a为原料进行本反应, 以54%收率获得2-二氟甲氧基苯腈.
在此基础上, 参考文献[12,26,39,40]报道, 提出了一种可能的机理(如Scheme 2所示). 首先, 氯二氟乙酸钠在碱及微波的作用下与肟羟基反应, 生成中间体5, 中间体5在微波加热条件下脱去二氧化碳及二氟卡宾, 生成1,2-苯并异噁唑中间体6. 中间体6在碱性条件下易脱去质子即发生Kemp消除生成中间体7. 中间体7与二氟卡宾(既来源于中间体5, 又来源于氯二氟乙酸钠)结合生成中间体8, 并立即接收质子生成产物3a. 在高温条件下, 中间体7也易接收质子生成2-羟基苯腈4a. 对于酮肟类底物, 反应生成中间体6时, 无活性质子, 无法进行后续转化.
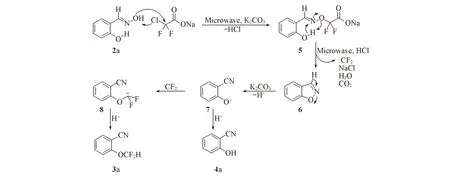
Scheme 2 Proposed reaction mechanism of coversion salicylaldehydeoxime to 2-(difluoromethoxy)benzonitrile
3 结 论
在300 W微波环境下, 以水杨醛肟类化合物为原料, 氯二氟乙酸钠为二氟甲基化试剂, 碳酸钾为碱, N,N-二甲基甲酰胺为溶剂, 经一锅脱水成腈及二氟甲基化反应制备了一系列2-二氟甲氧基苯腈类化合物. 该反应无需额外添加催化剂即可向化合物中同时引入二氟甲基和氰基, 具有反应迅速、 条件温和、 操作简便和实用性强的优点. 通过对溶剂、 碱、 二氟甲基化试剂、 微波功率、 反应温度和时间的筛选确定了最优反应条件; 通过含不同取代基水杨醛肟的扩展实验验证了其适用性; 并提出了可能的反应机理. 2-二氟甲氧基苯腈类化合物可用于芳基吡咯啶基甲酮类食欲素受体拮抗剂的合成[41], 在医药、 农药和材料等方面具有潜在的应用价值.
参 考 文 献
[1] Cheng Y., Guo A. L., Guo D. S., Curr. Org. Chem., 2010, 14, 977—999
[2] Sperry J. B., Sutherland K., Org. Process Res. Dev., 2011, 15(3), 721—725
[3] Fuchibe K., Koseki Y., Aono T., Sasagawa H., Ichikawa J., J. Fluorine Chem., 2012, 133, 52—60
[4] Sperry J. B., Farr R. M., Levent M., Ghosh M., Hoagland S. M., Varsolona R. J., Sutherland K., Org. Process Res. Dev., 2012, 16(11), 1854—1860
[5] Prakash G. K. S., Ganesh S. K., Jones J. P., Kulkarni A., Masood K., Swabeck J., Olah G. A., Angew. Chem. Int. Ed., 2012, 51(48), 12090—12094
[6] Levin V. V., Zemtsov A. A., Struchkova M. I., Dilman A. D., Org. Lett., 2013, 15(4), 917—919
[7] Liu G., Wang X., Xu X. H., Lu X., Tokunaga E., Tsuzuki S., Schibata N., Org. Lett., 2013, 15(5), 1044—1047
[8] Min Q. Q., Yin Z., Feng Z., Guo W. H., Zhang X., J. Am. Chem. Soc., 2014, 136(4), 1230—1233
[9] Matheis C., Jouvin K., Goossen L. J., Org. Lett., 2014, 16(22), 5984—5987
[10] Thomoson C. S., Wang L., Dolbier W. R. J., J. Fluorine Chem., 2014, 168, 34—39
[11] Huang Y., He X., Li H., Weng Z., Eur. J. Org. Chem., 2014, 33, 7324—7328
[12] Wang W., Hua M., Huang Y., Zhang Q., Zhang X., Wu J., Chem. Res. Chinese Universities, 2015, 31(3), 362—366
[13] Zhao Y., Huang W., Zheng J., Hu J., Org. Lett., 2011, 13(19) , 5342—5345
[14] Wang F., Huang W., Hu J., Chin. J. Chem., 2011, 29(12), 2717—2721
[15] Iida T., Hashimoto R., Aikawa K., Ito S., Mikami K., Angew. Chem. Int. Ed., 2012, 51(38), 9535—9538
[16] Dolbier W. R. J., Battiste M. A., Chem. Rev., 2003, 103(4), 1071—1098
[17] Leroux F., Jeschke P., Schlosser M., Chem. Rev., 2005, 105(3), 827—856
[18] Taylor E. C., Macor J. E., Pont J. L., Tetrahedro., 1987, 43(21), 5145—5158
[19] Mederski W. W. K. R., Osswald M., Dorsch D., Christadler M., Schmitges C. J., Wilm C., Bioorg. Med. Chem. Lett., 1999, 9(4), 619—622
[21] Firouzabadi H., Jamalian A., Tamami M., Iranpoor N., Lett. Org. Chem., 2006, 3(4), 267—270
[22] Sardarian A. R., Shahsavari-Fard Z., Shahsavari H. R., Ebrahimi Z., Tetrahedron Lett., 2007, 48(14), 2639—2643
[23] Tamilselvana P., Basavarajub Y. B., Sampathkumarc E., Murugesand R., Catal. Commun., 2009, 10(5), 716—719
[24] Ramón R. S., Bosson J., Díez-González S., Marion N., Nolan S. P., J. Org. Chem., 2010, 75(4), 1197—1202
[25] Davoodnia A., Khojastehnezhad A., Bakavoli M., Tavakoli-Hoseini N., Chinese J. Chem., 2011, 29(5), 978—982
[26] Whiting E., Lanning M. E., Scheenstra J. A., Fletcher S., J. Org. Chem., 2015, 80(2), 1229—1234
[27] Hegedüs A., Cwik A., Hell Z., Horváth Z., Esekc., Uzsokic M., Green Chem., 2002, 4, 618—620
[28] Ma D. L., Li X. L., Hao L., Zhang P. Z., Chen H., Zhao Y., Chem. J. Chinese Universities, 2014, 35(5), 959—964(马东来, 李小六, 郝乐, 张平竹, 陈华, 赵影. 高等学校化学学报, 2014, 35(5), 959—964)
[29] Kuang D. Z., Feng Y. L., Yu J. X., Zhang F. X., Jiang W. J., Peng Y., Zhu X. M., Tan Y. X., Chem. J. Chinese Universities, 2014, 35(8), 1629—1634(邝代治, 冯泳兰, 庾江喜, 张复兴, 蒋伍玖, 彭雁, 朱小明, 谭宇星. 高等学校化学学报, 2014, 35(8), 1629—1634)
[30] Li L., Wang F., Ni C., Hu J., Angew. Chem. Int. Ed., 2013, 52(47), 12390—12394
[31] Aldred R., Johnston R., Levin D., Neilan J., J. Chem. Soc. Perkin. Trans. 1, 1994, 13, 1823—1831
[32] Babua M. S. S., Krishnab P. G., Reddya K. H., Philip G. H., Main Group Chem., 2009, 8(2), 101—114
[33] Aakeröy C. B., Sinha A. S., Epa K. N., Spartz C. L., Desper J., Chem. Commun., 2012, 48(92), 11289—11291
[34] Kao C., Chen K., Journal of the Chinese Chemical Society, 1935, 3, 22—26
[35] Moghadam M., Tangestaninejad S., Mirkhani V., Mohammadpoor-Baltork I., Moosavifar M., Appl. Catal. A-Gen., 2009, 358(2), 157—163
[36] Gigant N., Claveau E., Bouyssou P., Gillaizeau I., Org. Lett., 2012, 14(3), 844—847
[37] Xu J., Wang X., Shao C., Su D., Cheng G., Hu Y., Org. Lett., 2010, 12(9), 1964—1967
[38] Shelke K. F., Spakal S. B., Shitole N. V., Shingate B. B., Shingare M. S., Org. Commun., 2009, 2(3), 72—78
[39] Ohsawa A., Kawaguchi T., Igeta H., Chem. Pharm. Bull., 1982, 30(12), 4352—4358
[40] McIntyre N. R., Lowe E. W. J., Merkler D. J., J. Am. Chem. Soc., 2009, 131, 10308—10319
[41] Martin B., Christoph B., Christine B., Markus G., Bibia H., Thierry S., Jodi T. W., [Ortho-Bi(hetero)aryl]-[2-(meta-bi(hetero)aryl)-pyrrolidin-1-yl]-methanone Derivatives as Orexin Receptor Antagonists and Their Preparation, WO 2014057435, 2014-04-17
(Ed.: P, H, W, K)
† Supported by the National Natural Science Foundation of China(Nos.21302071, 21471069), the Natural Science Foundation of Jiangsu Province, China(No.BK20130484) and the Scientific Research Foundation for Advanced Talents of Jiangsu University, China(No.12JDG089).
Microwave-assisted One-pot Conversion of Salicylaldehydeoximes to 2-(Difluoromethoxy)benzonitriles†
WANG Tao1, HUA Mingqing1*, LIU Weihan1, HUANG Yan1, ZHANG Qi1,2*
(1. Department of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China 2. Hainan Provincial Key Lab of Fine Chemistry, Hainan University, Haikou 570228, China)
Eight 2-(difluoromethoxy)benzonitrile derivatives including seven novel compounds were synthesized in moderate yields by one-pot multistep reaction of salicylaldehydeoximes using sodium 2-chloro-2,2-di-fluoroacetate(SCDA) as difluoromethylating reagent and potassium carbonate as base under microwave-assisted condition. The structures of all novel compounds were confirmed by nuclear magnetic resonance spectrum(1H NMR,13C NMR,19F NMR), infrared spectrum(IR) and high resolution mass spectrum(HRMS). The effects of difluorocarbene reagent, alkali, solvent, microwave power, reaction temperature and time on the reaction were considered. The optimized reaction conditions: salicylaldehydeoximes(2 mmol), 2-chloro-2,2-difluoroacetate(3 mmol), K2CO3(3 mmol) and N,N-dimethylformamide(2 mL), microwave power 300 W, reaction temperature 85 ℃, reaction time 20 min. A possible mechanism was proposed on the basis of the experiments and contrast tests.
Salicylaldehydeoxime; 2-(Difluoromethoxy)benzonitrile; Difluoromethylation; Microwave
10.7503/cjcu20150670
2015-08-21.
日期: 2016-01-07.
国家自然科学基金(批准号: 21302071, 21471069)、 江苏省自然科学基金(批准号: BK20130484)和江苏大学高级专业人才科研启动基金(批准号: 12JDG089)资助.
O625.6
A
联系人简介:华明清, 男, 博士, 讲师, 主要从事有机合成研究. E-mail: huamq840710@163.com
张 岐, 男, 博士, 教授, 博士生导师, 主要从事有机合成和磁共振成像造影剂等方面的研究.
E-mail: qzhang@ujs.edu.cn
猜你喜欢
能源与环境(2022年5期)2023-01-10
绿色科技(2020年14期)2020-12-23
化学工程师(2020年5期)2020-06-30
铜仁学院学报(2018年6期)2018-07-05
中成药(2017年4期)2017-05-17
中成药(2017年3期)2017-05-17
军事文摘·科学少年(2016年10期)2016-12-08
军事文摘(2016年20期)2016-11-07
合成化学(2016年1期)2016-02-25
应用化工(2014年10期)2014-08-16
