
中国桑属15个种RAD-seq高通量测序*
2016-11-29 05:58:10陈祥平柯皓天刘凯旋王茜龄任艳红陈仁芳
蚕学通讯 2016年3期
陈祥平 吕 银 刘 玲 柯皓天 刘凯旋 王茜龄 任艳红 陈仁芳*
(1.四川省丝绸科学研究院,成都 610031;2.西南大学生物技术学院,重庆 400716)
中国桑属15个种RAD-seq高通量测序*
陈祥平1吕 银1刘 玲1柯皓天1刘凯旋1王茜龄2任艳红2陈仁芳2*
(1.四川省丝绸科学研究院,成都 610031;2.西南大学生物技术学院,重庆 400716)
利用RAD-seq对中国桑属15个种进行了高通量测序,总共得到36.72Gb clean data,总Tags数2 788 927(reads) ,平均每个种原始数据都在10M以上,Tags都在20万条以上,质量值Q30都在90%以上。用Stacks软件对15个种进行比对,获得68904个SNPs位点。用最大似然法建树,分支图首先将白桑、广东桑分出,接着是山桑、鲁桑、瑞穗桑,再次分出的是鸡桑、细齿桑、蒙桑和鬼桑,最后分出的是黑桑、川桑、华桑、滇桑、长穗桑、奶桑。分支图能将栽培种和野生种完全分开;可以将蒙桑和鬼桑、鸡桑、华桑、川桑、奶桑分开。认为白桑、广东桑属原始类型,长穗桑、奶桑属进化类型;山桑、鲁桑、瑞穗桑这三个种被分在一个分支,自检支持率99%,黑桑、川桑这两个种被分在一个分支,自检支持率56%,长穗桑、奶桑这两个种被分在一个分支,自检支持率100%,说明这些种之间有较近的亲缘关系,桑属RAD-seq测序能大规模筛查SNPs位点,系统发育分析的准确性就更加可靠。
中国桑属;RAD-seq;SNP标记;系统发育
桑属(Morus)为桑科的模式属,由Linne(1753)建立[1],其后虽有许多学者进行研究[2-12],但由于学者们对物种的认识不同,分出的种数差异很大。桑属的分子系统学也先后有许多学者进行研究[13-21]。但这些研究由于所用的方法、DNA片段,信息位点有限,不能将所有桑种分开。
限制性内切酶位点相关DNA(Restriction-site Associated DNA, RAD)由Miller等[22]2007年提出。它是在新一代测序技术基础上发展起来的一种DNA新技术。该技术利用限制性内切酶对基因组进行酶切, 产生一定大小的片段, 构建测序文库, 对酶切后产生的RAD标记进行高通量测序[23]。由于RAD标记代表整个基因组特异性酶切位点附近的小片段DNA标签,又由于新一代测序技术通量高。因此,通过对RAD标记测序能够获得成千上万的单核苷酸多态性(Single nuc-leotide polymorphism, SNP)标记[24-26]。RAD-seq 已成功应用于生物SNP标记的开发、超高密度遗传图谱的构建、动植物重要经济性状的QTL定位、辅助全基因组重测序等研究领域[27-33],在群体遗传结构、系统演化分析方面也在温带竹子、美国橡树、北美瓶草蚊、马先蒿、蝶属、三刺鱼上有研究[34-39],但未见在桑属上报道。因此,测定了中国桑属15个种的RAD-seq序列 ,进行SNPs开发和系统进化分析。现将研究结果报道如下。
1 材料与方法
1.1 材料
材料取中国桑属15个种,每个种取1-5位嫩叶后立即装入冰盒,带回实验室-70℃冰箱保存备用,各个种采集地、纬度、经度、海拔(表1)。

表1 桑属采样信息
1.2 实验流程
(1)提取每个样品基因组DNA;
(2)采用限制性内切酶EcoRⅠ(GAATTC)酶切: 0.1-1ug基因组DNA在50uL体系中用20u的EcoRI于37℃消化15min,65℃20min使内切酶失活;
(3)连接P1接头:在消化后的DNA片段两端加P1 Adapter,再次65℃20min使连接酶失活;
(4)片断化:带有不同P1 接头的样品混合在一起,采用物理方法打断成300-500bp的片段,1%琼脂糖凝胶电泳后回收300-700bp的DNA, 末端平化后加A;
(5)连接P2接头: P2 adapter 是一个发散型的“Y” adapter, 它可以防止缺乏P1接头的基因组片段扩增,即只有两端接头种类不同的片段才能被选择性扩增;
(6)PCR扩增、纯化、上机测序:取5uL,用P1和P2引物PCR扩增, 18个循环后,跑胶纯化回收300-500bpDNA片段,上机测序,测序平台Illumina hiseq4000,测序方法Illumina/Solexa 聚合酶合成测序,测序深度3X,测序在华大基因进行。
1.3 序列分析
(1)序列过滤
raw reads包含低质量序列、adapter序列等,需要经过一系列数据处理来,得到clean data,详细步骤如下:去掉含有adapter的reads;去掉低质量的数据(过滤参数为质量值低于12占整条reads的40%或者以上,删除整条reads;去除含N(表示无法确定碱基信息)比例大于3%的reads;去除序列5’端未包含样本barcode(4-8bp)信息的reads(一个样品对应一个barcode);剪切掉5′端barcode、酶切识别位点。如果剪切掉barcode的reads 5’端未含酶切识别位点,则去除该reads。
(2)用Stacks软件[40]对reads进行整理,通过ustacks→cstacks→sstacks→population程序处理,获得SNPs位点。
(3)系统发育树构建:population步程序得到的phylip文件上传到Cipres Science Gateway网站(https://www.phylo.org/),RAxML-HPC BlackBox程序(Phylogenetic tree inference using maximum likelihood最大似然法)建树。
2 结果
2.1 数据产出
经过上述过滤raw data处理之后,总共得到36.72Gb clean data,总Tags数2 788 927(reads),并根据个体barcode区别,将过滤后的reads细分至每个个体,数据产出(表2)。
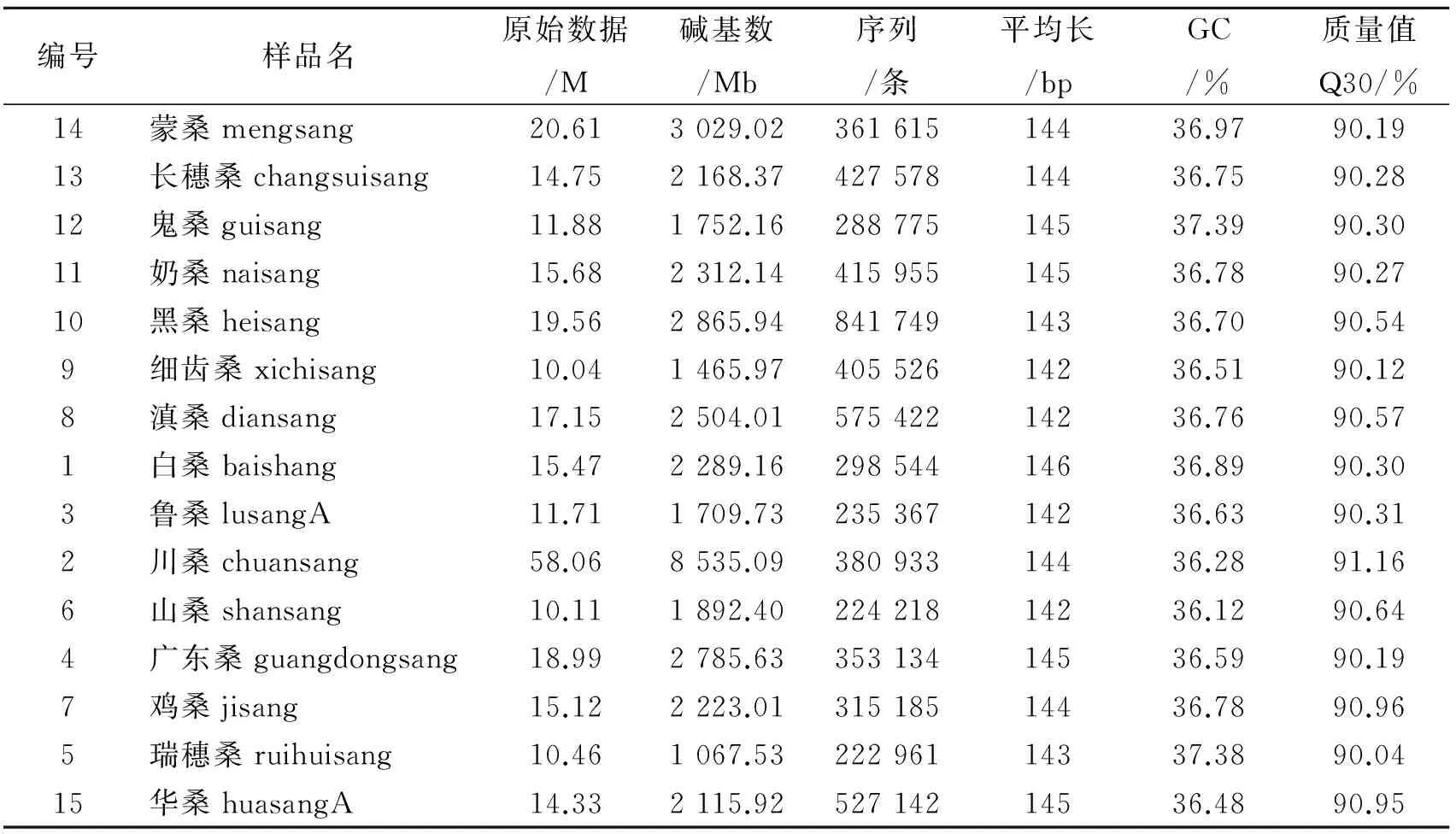
表2 数据产出统计
从表2可以看出每个种原始数据都在10M以上,通过过滤raw data,用Stacks软件处理后得到序列Tags都在20万条以上,质量值Q30都在90%以上,且每个种得到Tags数量都较均匀,均可用于后续SNP标记检测和系统发育分析。
2.2 SNP标记的检测
用Stacks软件对reads进行整理,通过population程序,对15种进行比对,获得68 904个SNPs位点,为后续的系统发育分析的准确性创造了条件。
2.3 系统发育分析
用Stacks软件population程序得到的phylip文件,上传到Cipres Science Gateway网站(https://www.phylo.org/),启动RAxML-HPC BlackBox程序,用最大似然法建树,得到中国桑属15个种的系统发育树(图3)。
从图3可以看出,白桑、广东桑首先分出,接着是山桑、鲁桑、瑞穗桑,再次分出的是鸡桑、细齿桑、蒙桑和鬼桑,最后分出的是黑桑、川桑、华桑、滇桑、长穗桑、奶桑。山桑、鲁桑、瑞穗桑这三个种被分在一个分支,自检支持率99%,黑桑、川桑这两个种被分在一个分支,自检支持率56%,长穗桑、奶桑这两个种被分在一个分支,自检支持率100%。

图1 基于RAD-seq中国桑属15个种系统发育(分支间的数据为50%以上的自检支持率)
3 讨论
3.1 桑属RAD-seq测序要求
RAD-seq属新一代高通量测序,对DNA质量要求较高,样品浓度:100-200ng/μl,样品纯度:OD260/280=1.8-2.0。因此,采样要用冰盒采集鲜样,尽量采嫩叶,立即送回实验室-70℃冷藏保存,DNA提取最好用Plant Genomic DNA Kit。另外,本次研究主要是开发桑属SNPs,进行系统进化分析,要求测序深度至少3X,Q30至少90%以上。
3.2 桑属RAD-seq测序能大规模筛查SNPs位点
本研究采用Illumina hiseq4000测序平台, Illumina/Solexa聚合酶合成测序,测序深度3X,对中国桑属15个种进行RAD-seq测序,过滤掉一些低质量的reads,用Stacks软件,population程序,对15种进行比对,获得68 904个SNPs位点,相比用常规方法(ITS序列116个SNPs位点,ITS、trnL-F、rps16三个片段合并261个SNPs位点[18-19]),SNPs位点可成万倍的增加,后续的系统发育分析的准确性就更加可靠。
3.3 基于RAD-seq中国桑属15个种系统发育树的几个新观点
本文根据筛查到的68 904个SNPs位点,用最大似然法建立的系统发育树有几个新观点提供给读者商榷。
(1) 分支图可以将栽培种和野生种完全分开;
(2) 可以将蒙桑和鬼桑、鸡桑、华桑、川桑、奶桑分开(常规用ITS、trnL-F、rps16序列不能将蒙桑和鬼桑、鸡桑与白桑分开,不能将华桑、川桑、奶桑分开);
(3) 白桑、广东桑属原始类型,长穗桑、奶桑属进化类型;
(4) 山桑、鲁桑、瑞穗桑亲缘关系近,黑桑、川桑亲缘关系近,长穗桑、奶桑亲缘关系近,细齿桑与蒙桑、鬼桑亲缘关系近;
(5) 中国植物志将山桑定为白桑的变种[41],理由是山桑无自然地理分布,而本研究将山桑、鲁桑、瑞穗桑分在一个分支,自检支持率99%,鲁桑、瑞穗桑也无自然地理分布。因此,认为鲁桑、瑞穗桑不作种级单位为宜;
(6) 滇桑与长穗桑、奶桑亲缘关系近,且滇桑为长穗类,应为长穗类里的一个种,而不作为蒙桑的变种。
3.4 本研究需要进一步改进之处
(1)由于川桑全基因已测序,应根据川桑全基因信息筛选合适的内切酶进行酶切建库,有目的的筛查SNPs位点;
(2)stacks软件应选pstacks程序,而不是选ustacks程序,因为桑属已有川桑全基因组信息;
(3)每个种应取更多样,在population步,统计分析桑群体遗传学Pi、Fis、Fst等相关数据。
[1] LINNE C V.1753.Species plantarum[M]. 2:986.
[2] MORETTI G.Prodromo di una monografia delle specie del centreMorus[M].1842:564.
[3] SERINGE N C.Description,culture et taille des murierrs[M].1855(98):423-425.
[4] BUREAU E.MORACEAE.Prodromus systematis naturalis regni vegetabilis.Volume 17 XVIL[M].AIph.de Candolle.(ed.).Paris, France.1873:211-288.
[5] SCHNEIDER C K.lantae Wilsonianae[J].Sarg.PI.Wils.1916,3(2):296-297.
[6] KOIDZUMI G.Taxonomical discussion on Morus plants[J].Bull Imp Sericult Exp Stat,1917(3):1-62.
[7] 陈嵘.中国树木分类学[M].北京:科学出版社,1937:228-231.
[8] 胡先骕.植物分类学简编[M].北京:科学技术出版社,1957:56.
[9] 张秀实.桑科新分类群[J].植物分类学报,1984,22(1):64-76.
[10]吴征镒,张秀实.中国桑科的一些新分类单位[J].云南植物研究,1989,11(1):24-34 .
[11]曹子余.中国桑属(桑科)新分类群[J].植物分类学报,1991,29(3):264-267.
[12]曹子余.中国植物志资料(桑科) [J].云南植物研究, 1995,17(2):153-154,158.
[13]向仲怀,张孝勇,余茂德,等.采用随机扩增多态性DNA技术(RAPD)在桑属植物系统学研究的应用初报[J].蚕业科学,1995(4):203-208.
[14]冯丽春,杨光伟,余茂德,等.利用RAPD对桑属植物种间亲缘关系的研究[J].中国农业科学,1997,30(1):52-56.
[15]杨光伟,冯丽春,张孝勇,等.桑属种群遗传结构变异分析[J].蚕业科学,2003,29(4):323-329.
[16]赵卫国, 潘一乐, 张志芳.桑属植物ITS序列研究与系统发育分析[J].蚕业科学,2004,30(1):11-14.
[17]汪伟,王兴科,朱昱萍.基于trnL内含子序列的桑属植物分子系统学初报[J].蚕业科学,2008,34(2):89-103.
[18]陈仁芳,余茂德,刘秀群,等.桑种质资源ITS序列与系统进化分析[J].中国农业科学,2010,43(8):34-42.
[19]陈仁芳,张泽,唐洲,等.桑属ITS、trnL-F、rps16序列与进化分析[J].中国农业科学,2011,44(8):1553-1561.
[20]MADHAV P.Systematics and Reproductive Biology of the GenusMorusL.(Moraceae)[D].Kansas:Kansas State University,2008.
[21]MADHAV P NEPAL,CAROLYN J FERGUSON.Phylogenetics of Morus(Moraceae) Inferred from ITS and trnL-trnF Sequence Data[J].Systematic Botany, 2012,37(2):442-450.
[22]MILLER M R,DUNHAM J P,AMORES A,et al.Ranid and costeffective polvmornhism identification and genotvping using restriction site associated DNA(RAD)markers[J].Genome Res,2007,17(2):240-248.
[23]H.C.ROWE, S.RENAUT,A.GUGGISBERG.RAD in the realm of next-generation sequencing technologies[J].Molecular Ecology,2011,20:3499-3502.
[24]NATHAN A.BAIRD, PAUL D.ETTER, TRESSA S.ATWOOD, et al.Rapid SNP discovery and geneticmapping using sequenced RAD markers[J].PlosOne,2008,3(10):e3376.
[25]VAN TASSELL CP, SMITH TP, MATUKUMALLI LK, et al.SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries[J].Nat Methods,2008,5(3):247-252.
[26]J.M.PUJOLAR,M.W.JACOBSEN,J.FRYDENBER G, et al.A resource of genome-wide single-nucleotide polymorphisms generated by RAD tag sequencing in the critically endangered European eel[J].Molecular Ecology Resources,2013,13:706-714.
[27]JOHN W.DAVEY,MARKL.BLAXTER.RADSeq:next-generation population Genetics[J].Briefings in Functional Genomics,2011,9(5):416-423.
[28]ADAM D.L EACHÉ,BARBARA L.BANBURY,JOSEPH FELSENSTEIN,et al.Short Tree, Long Tree, Right Tree, Wrong Tree:New Acquisition Bias Corrections for Inferring SNP Phylogenies[J].Systematic Biology Advance,2015,00(0):1-16.
[29]岳桂东, 高强, 罗龙海, 等.高通量测序技术在动植物研究领域中的应用[J].中国科学:生命科学,2012,42(2):107-124.
[30]HOHENLOHE PA, AMISH JS, CATCHEN MJ, et al.Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout[J]. Mol Ecol Resour,2011,11(Suppl.1):117-122.
[31]PFENDER WF, SAHA MC, JOHNSON EA, et al.Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne[J].Theor Appl Genet,2011,122(8):1467-1480.
[32]POLAND JA, BROWN PJ, SORRELLS ME, et al.Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach[J].PlosOne,2012,7(2):e32253.
[33]刘艳玲.莲野生居群遗传多样性评价及高密度遗传连锁图谱的构建[D].武汉:华中农业大学,2013.
[34]ANDREW L.HIPP, DEREN A.R.EATON, JEANNINE CAVENDER-BARES, et al.A Framework Phylogeny of the American Oak Clade Based on Sequenced RAD Data[J].PlosOne,2014,9(4):1-12.
[35]X.Q.WANG,L.ZHAO,D.A.R.EATON,et al.Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae:Bambusoideae) using RAD sequencing[J].Molecular Ecology Resources,2013,13:938-945.
[36]JULIAN CATCHEN,SUSAN BASSHAM,TAYLOR WILSON,et al.The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing[J].Molecular Ecology,2013,22:2864-2883.
[37]KEVIN J.EMERSON, CLAYTON R.MERZ, JULIAN M.CATCHEN,et al.Resolving postglacial phylogeography using high-throughput sequencing[J].PNAS,2010,107(31):16196-16200.
[38]DEREN A.R.EATON,RICHARD H.REE.Inferring Phylogeny and Introgression using RADseq Data:An Example from Flowering Plants (Pedicularis:Orobanchaceae)[J].Syst.Biol,2013,62(5):689-706,
[39]NICOLA J.NADEAU, SIMON H.MARTIN, KRZYSZTOF M.KOZAK, et al.Genome-wide patterns of divergence and gene flow across a butterfly radiation[J].Molecular Ecology,2013,22(3):814-826.
[40]CATCHEN J,HOHENLOHE P A,BASSHAM S,et al.Stacks:an analysis tool set for population genomics[J].Molecular Ecology,2013,22:3124-3140.
[41]中国植物志编辑委员会.中国植物志23卷[M].北京:科学出版社,1998:2-23.
RAD-seq High-Throughput Sequencing of 15 Species ofMorus
CHEN Xiang-ping1LÜ Yin1LIU Ling1KE hao-tian1LIU Kai-xuan1WANG Xi-ling2REN Yan-gong2CHEN Ren-fang2*
(1.TheSilkResearchInstituteofSichuanProvince,Chengdu610031,China;2.CollegeofBiotechnology,SouthwestUniversity,Chongqing400716,China)
In this study, RAD-seq (restriction site-associated DNA sequencing) was performed toidentify the SNP loci between 15 species of the genusMorus. A total of 36.72 Gb clean data were sequenced by Illumina hiseq 4000 and a total of 68904 SNP loci were obtained by Stacks software. The data sets were analysed using the maximum-likelihood method. The cultivated and wild species were completely separate,M.albaandM.atropurpureawere original genera of mulberry, however,M.wittiorumandM.macrourawere of the evolutionary type.M.serrata,M.mongolica, andM.mongolicavar.diabolicahad close genetic relationship,whileM.yunnanensis,M.wittiorumandM.macrourahad close genetic relationship. Analyses based on these RAD tags yielded robust phylogenetic inferences, even with data set constructed from surprisingly few loci. The study illustrates the potential for resolving difficult phylogenetic relationships in genusMorus.
Morus; Restriction site-associated DNA; SNP marker; Phylogeny
* 资助项目:四川省科技厅应用基础研究计划项目。通讯作者:陈仁芳,博士,副教授,从事桑树学研究。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
传染病信息(2021年6期)2021-02-12 01:52:58
生物学通报(2019年3期)2019-02-17 18:03:58
中国篆刻·书画教育(2017年5期)2017-06-08 00:42:41
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
化学工业与工程(2015年1期)2015-02-10 03:01:41
赤峰学院学报·自然科学版(2013年4期)2013-07-31 22:01:40
