
一测多评法测定新疆紫草中主要萘醌类成分的含量
2016-11-19 12:54赵文文吴智敏吴霞赵海誉陈筱清
中国中药杂志 2016年20期
关键词:萘醌
赵文文+吴智敏+吴霞+赵海誉+陈筱清


[摘要]以β,β′-二甲基丙烯酰阿卡宁为内参物,测定新疆紫草中乙酰紫草素、β-乙酰氧基异戊酰阿卡宁、异丁酰紫草素、α-甲基-正丁酰紫草素的含量,建立紫草中5 种萘醌类成分的一测多评定量分析方法。采用高分辨LC-MS对含量测定的5个色谱峰进行鉴定。采用不同高效液相(HPLC)系统和不同色谱柱考察相对校正因子的耐用性,同时采用外标法与一测多评法对16 批新疆紫草药材进行分析,并对含量测定结果进行比较。结果表明相对校正因子的耐用性良好,16 批样品一测多评法与外标法所得结果无显著差异。所建立的含量测定方法可用于同时测定新疆紫草中5种萘醌类成分。
[关键词]新疆紫草; 一测多评; 相对校正因子; 萘醌; LC-MS
[Abstract]This study is to determine five naphthaquinones (acetylshikonin, β-acetoxyisovalerylalkannin, isobutylshikonin, β,β′-dimethylacrylalkannin,α-methyl-n-butylshikonin) by quantitative analysis of multi-components with a single marker (QAMS). β,β′-Dimethylacrylalkannin was selected as the internal reference substance, and the relative correlation factors (RCFs) of acetylshikonin, β-acetoxyisovalerylalkannin, isobutylshikonin and α-methyl-n-butylshikonin were calculated. Then the ruggedness of relative correction factors was tested on different instruments and columns. Meanwhile, 16 batches of Arnebia euchroma were analyzed by external standard method (ESM) and QAMS, respectively. The peaks were identifited by LC-MS. The ruggedness of relative correction factors was good. And the analytical results calculated by ESM and QAMS showed no difference. The quantitative method established was feasible and suitable for the quality evaluation of A. euchroma.
[Key words]Arnebia euchroma; quantitative analysis of multi-components by single marker (QAMS); relative correction factor (RCF); naphthaquinone; LC-MS
doi:10.4268/cjcmm20162014
紫草始载于《神农本草经》,味苦,性寒,具有凉血、活血、解毒和透疹的功能[1]。紫草中主要有效成分为羟基萘醌类,具有抗菌、抗肿瘤、抗病毒、抗炎、抗过敏、保肝降酶等作用[2]。《中国药典》2015年版(一部)收载紫草来源于紫草科植物新疆紫草Arnebia euchroma (Royle) Johnst. 或内蒙紫草A. guttata Bunge 的干燥根,以β,β′-二甲基丙烯酰阿卡宁为含量测定指标[3]。近年来已有文献报道紫草的多成分含量测定[4-6],但采用一测多评法进行紫草的质量评价未见报道。本试验建立一测多评方法,以β, β′-二甲基丙烯酰阿卡宁为内参物,同时测定乙酰紫草素,β-乙酰氧基异戊酰阿卡宁,异丁酰紫草素,α-甲基-正丁酰紫草素的含量,为紫草的质量评价提供参考。
1 材料
Agilent 1200 HPLC 系统(美国Agilent 科技有限公司),岛津 LC-20A HPLC 系统(日本岛津公司),Waters e2695 HPLC 系统(美国沃特世科技有限公司),双压线性离子阱串联高分辨质谱Orbitrap Velos Pro (Thermo Fisher公司),戴安Ultimate 3000 超高效液相色谱仪,KQ-500VDE 型舒美双频数控超声波清洗器(昆山市超声仪器有限公司),OHAUS DV215CD 1/10万分析天平(美国奥豪斯仪器有限公司)。
甲醇、乙腈为色谱纯(美国Fisher公司),甲酸为分析纯(北京益利精细化学品有限公司),氨水为分析纯(国药集团化学试剂有限公司),纯净水(娃哈哈)。
乙酰紫草素对照品(上海士锋生物科技有限公司,批号:B11181),β-乙酰氧基异戊酰阿卡宁对照品(上海源叶生物科技有限公司,批号:B21977),异丁酰紫草素对照品(上海源叶生物科技有限公司,批号:B21519),β,β′-二甲基丙烯酰阿卡宁对照品(中国食品药品检定研究院,批号:111689-201504),α-甲基-正丁酰紫草素对照品(东京化成工业株式会社,批号:M1028)。
16批紫草药材(1#,2#,3#,4#,5# 购自安徽亳州中药材市场,6#,7#,8#,9#,10#,11#,12#,13#,14#,15#,16# 购自河北安国中药材市场),产地均为新疆。经首都医科大学中医药学院李佳教授鉴定为新疆紫草A. euchroma的干燥根。
2 方法与结果
2.1 一测多评法的建立
2.1.1 色谱条件 含量测定采用Hibar Purospher STAR LP RP-18e(4.6 mm×250 mm, 5 μm)色谱柱,柱温为25 ℃,检测波长275 nm,流动相为乙腈-0.05%甲酸水(75∶25)等度洗脱,流速1.0 mL·min-1,进样量10 μL,见图1。
液质联用色谱分离采用Thermo Hypersil BDS C18(2.1 mm×150 mm, 2.4 μm)色谱柱,柱温为30 ℃,样品盘温度4 ℃,流动相为乙腈-0.2%氨水(75∶25)等度洗脱,混合对照品溶液进样1 μL,样品溶液进样1 μL。
电喷雾电离源,负离子扫描模式,加热器温度为350 ℃,毛细管温度为350 ℃,毛细管电压为35 V,喷雾电压为3.5 kV,鞘气(N2)流速为35 arb,辅助气(N2)流速为10 arb,样品在FT模式下进行全扫描(分辨率R为3万,扫描范围为m/z50~1 000)。数据采集和分析采用Xcalibur软件。
2.1.2 对照品溶液的制备 分别取乙酰紫草素,β-乙酰氧基异戊酰阿卡宁,异丁酰紫草素,β, β′-二甲基丙烯酰阿卡宁,α-甲基-正丁酰紫草素适量,精密称定,用甲醇溶解,制成1 L含上述5个成分分别为294, 599, 162, 198, 588 mg的混合对照品储备液。再将对照品储备液稀释 2, 5, 10, 50, 100, 500倍得到共7个梯度浓度的混合对照品溶液。
2.1.3 供试品溶液的制备 取样品粉末0.5 g,精密称定,置于50 mL具塞锥形瓶中,加甲醇50 mL,称重,超声处理(功率250 W,频率33 kHz)30 min,放冷,称重,用甲醇补足失重,滤过,取续滤液,即得。
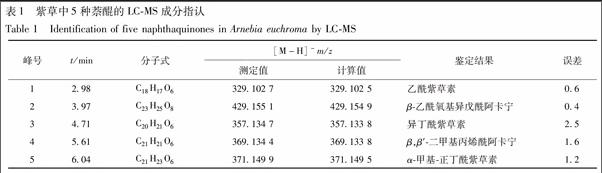
2.1.4 LC-MS 成分确认 采用LC-MS法按2.1.1项色谱及质谱条件,对含量测定的5个色谱峰所对应的成分进行鉴定,通过与混合对照品溶液中5个成分准分子离子峰对照,确认供试品溶液中与混合对照品溶液中相同保留时间色谱峰,分别为乙酰紫草素、β-乙酰氧基异戊酰阿卡宁、异丁酰紫草素、β,β′-二甲基丙烯酰阿卡宁和α-甲基-正丁酰紫草素,见表1。
2.1.5 线性关系考察 分别精密吸取2.1.2项下7个混合对照品溶液进样分析,以峰面积为纵坐标,进样浓度(mg·L-1)为横坐标,绘制标准曲线,得到紫草中上述5个成分的线性方程见表2。
2.1.6 相对校正因子计算[7] 以β, β′-二甲基丙烯酰阿卡宁为内参物,按公式fmn = fm/fn= (Wm/Am)/(Wn/An),其中Wm/Am,Wn/An分别表示内参物和待测化合物单位浓度的响应值。计算不同浓度(n=7)其余4个化合物的相对校正因子,结果fDAL/AS,fDAL/AAL,fDAL/IBS和fDAL/MBS平均值分别为0.99,1.40,0.85,1.23,RSD为0.50%,0.20%,2.2%,1.1%。
2.1.7 精密度试验 精密量取同一浓度混合对照品溶液,连续进样6次,记录峰面积,5个萘醌类成分的峰面积RSD依次为0.36%,0.29%,0.80%,0.41%,0.27%。表明仪器精密度良好。
2.1.8 稳定性试验 取同一供试品溶液,分别于配制后的0,2,4,8,12,24 h进样分析,记录峰面积,乙酰紫草素、β-乙酰氧基异戊酰阿卡宁,异丁酰紫草素,β,β′-二甲基丙烯酰阿卡宁,α-甲基-正丁酰紫草素的峰面积RSD值分别为1.8%,1.2%,2.2%,1.2%,1.4%。表明样品在24 h内稳定。
2.1.9 重复性试验 精密称取同一样品粉末,按2.1.3项下方法平行制备6份溶液,按2.1.1项下色谱条件进样分析,根据标准曲线计算各成分的含量,计算RSD分别为1.5%,1.7%,2.0%,1.5%,1.9%。表明方法重复性良好。
2.1.10 加样回收率试验 精密称取同一批次样品 0.25 g共9份,分别加入一定量(高、中、低各3份)的对照品溶液,按2.1.3项下操作,计算加样回收率,5个萘醌类成分加样回收率平均值依次为97.40%,96.60%,99.90%,99.50%,96.30%,RSD依次为1.6%,2.9%,3.0%,1.4%,2.9%。表明方法准确度良好。
2.2 一测多评法的耐用性
2.2.1 相对较正因子的耐用性 试验选用Agilent 1200,岛津LC-20A和Waters e2695 3套高效液相色谱系统,Hibar Purospher STAR LP RP-18e(4.6 mm×250 mm, 5 μm),Kromasil C18(4.6 mm×250 mm, 5 μm),Grace Vision HT C18-HL(4.6 mm×250 mm, 5 μm),YMC-Pack ODS-AQ(4.6 mm×250 mm, 5 μm),Agilent Zorbax XDB-C18(4.6 mm×250 mm, 5 μm)5种色谱柱,考察不同色谱系统对相对校正因子的影响,见表3。
2.2.2 待测组分色谱峰的定位 采用相对保留值作进行色谱峰定位,考察不同色谱系统对相对保留 时间的影响,见表4。
2.2.3 一测多评法与外标法结果比较 将收集的16批新疆紫草按2.1.3项下方法制备样品溶液,按2.1.1项下条件进行分析,采用外标法和QAMS法分别计算5种萘醌成分的含量,以5个成分之和计算总萘醌的含量,见表5,图2。
3 结果与讨论
3.1 提取方法考察
根据紫草萘醌类成分的性质,分别对提取溶媒(甲醇、丙酮、石油醚)、提取方法(超声、回流、冷浸) 与提取时间(20,30,40 min)进行了考察,确定甲醇超声提取30 min,该方法简便且药材中的萘醌类成分提取较完全。将该方法与《中国药典》2015年版(一部)紫草含量测定项下样品溶液进行比较,发现所测得含量无明细差异,考虑到方法的的简便性,仍采用甲醇超声提取法。
3.2 内参物的确定
从对照品来源有保证,廉价、易得方面考虑,选择β,β′-二甲基丙烯酰阿卡宁为内参物,体现一测多评法简便、易操作、低成本的特点。
3.3 相对校正因子考察
本实验考察了3套HPLC 系统、5种色谱柱对相对校正因子的影响,结果表明色谱系统对相对保留因子影响较小,RSD均<3%,重复性良好。5个萘醌类成分具有相同的结构母核,仅存在异己烯侧链的取代基不同,但其中3个成分校正因子偏离1较多,主要因为异己烯侧链作为紫外吸收助色团,取代基不同,直接影响化合物的摩尔吸光系数,此外,侧链取代基不同直接影响分子摩尔质量,导致不同化合物单位质量浓度(mg·L-1)中摩尔浓度(mol·L-1)不同,也是校正因子偏离1较多的主要原因之一。
3.4 色谱峰的定位
本试验通过相对保留时间来评价色谱峰的定位效果,考察不同色谱系统对相对保留时间的影响,除Grace Vision HT色谱柱对乙酰紫草素相对保留时间影响较大外,其他色谱柱及HPLC系统对相对保留时间影响均较小,RSD均<5%,并且5个成分出峰顺序均不受色谱系统的影响,见表4。
3.5 一测多评法与外标法结果比较
本研究首次建立一测多评法测定新疆紫草中主要活性成分含量。各成分含量测定结果显示一测多评法与外标法无显著性差异,提示建立的相对校正因子具有较好的可信度,QAMS计算结果可靠。
3.6 含量测定
目前,我国尚未见紫草栽培的报道,野生资源较为紧张。收集到的16批新疆紫草中,药典含量测定指标成分β, β′-二甲基丙烯酰阿卡宁仅在4#,14#,15#样品中符合不少于0.30%的要求。该成分在总萘醌中所占比例较低,而在某些混伪品中,该成分含量较高[8],这也是市场上出现较多混伪品及掺杂现象的主要原因之一。因此,同时检测多个萘醌类成分含量更有利于评价紫草药材质量的优劣。不同批次新疆紫草萘醌含量差异较大,以5个成分之和计算总萘醌的量在2.71~49.68 mg·g-1,其中α-甲基-正丁酰紫草素含量与总萘醌含量呈正相关,见图2,能够一定程度反映药材中总萘醌含量的高低,对紫草药材质量评价具有重要意义。
[参考文献]
[1]阴健,郭力弓.中药现代研究与临床应用[M].北京:学苑出版社,1993.
[2]赵雪梅,邓文,李莹,等.紫草不同提取物抗炎及抑菌作用实验研究[J].时珍国医国药,2008, 19(7):1603.
[3]中国药典.一部[S].2015:340.
[4]阿呷尔布,聂丽娟,卓玛东智,等.多指标成分定量结合指纹图谱分析评价不同来源藏紫草的质量[J].中国中药杂志,2015,40(22):4442.
[5]郝鹤,李鹏跃,叶和春,等.新疆紫草7种萘醌类成分的同时测定[J].中国实验方剂学杂志,2013,19(18):108.
[6]陈璐璐,周若龙,杨柳,等. 紫草中萘醌类化合物的质谱鉴别及3种成分的UPLC同时测定[J]. 中药新药与临床药理,2012,23(1):77.
[7]王智民,高慧敏,付雪涛,等.一测多评法中药质量评价模式方法学研究[J].中国中药杂志, 2006, 31 (23):1925.
[8]Cheng M, Mo Q G, Xu Y, et al. Quality evaluation of Arnebiae Radix using multiple qualitative and quantitative methods coupled with multivariate statistical analysis[J].Curr Pharm Anal, 2013(9):217.
[责任编辑 丁广治]
猜你喜欢
食品工业(2022年11期)2022-11-28
食品与发酵工业(2022年5期)2022-03-30
广州化学(2020年6期)2020-12-28
当代化工(2020年5期)2020-08-25
中国民族民间医药(2020年4期)2020-05-07
黑龙江八一农垦大学学报(2020年2期)2020-05-06
化工设计通讯(2016年11期)2016-02-08
石河子大学学报(自然科学版)(2011年1期)2011-10-14
化学与粘合(2011年2期)2011-09-24
