
6-乙烯氧基-5,12-萘并萘醌光致变色性能理论研究
2016-02-08 03:15:18尹京花
化工设计通讯 2016年11期
尹京花
(延边大学理学院化学系,吉林延吉 133002)
6-乙烯氧基-5,12-萘并萘醌光致变色性能理论研究
尹京花
(延边大学理学院化学系,吉林延吉 133002)
用密度泛函理论的B3LYP/6-31G 方法和从头算的CIS/6-31G方法分别研究了6-乙烯氧基-5,12-萘并萘醌基态和激发态的异构化反应。反应势能面显示,6-乙烯氧基-5,12-萘并萘醌在光异构化反应中基态和激发态不能构成四能级反应,热力学上有利于以trans醌式构型存在。此外,用TD/B3LYP方法在溶剂存在下计算了上述化合物的紫外吸收光谱,计算所得到的光谱数据与实验值基本一致,与光异构化反应的光激发条件相符合。
萘并萘醌;光致变色;光异构化;密度泛函理论
光致变色是指在光作用下一个化合物异构化为其异构体,之后在光或热的作用下异构体又变回起始化合物。光致变色材料广泛应用在信息存储材料、传感器、智能开关等领域[1-4]。众多光致变色材料中,萘并萘醌类化合物因为其良好的耐疲劳性和热稳定性而引起关注[5]。为了研究迁移基团结构对其变色性的影响,探明萘并萘醌化合物光致变色机理,本文选用以乙烯基作为迁移基团的萘并萘醌衍生物,研究了萘并萘醌衍生物中迁移基团的分子结构对母体萘醌环共轭程度以及在光致变色过程中的作用,以此为萘并萘醌光致变色材料的选取提供理论支持。
1 计算方法
用密度泛函的B3LYP/6-31G方法对乙烯基取代的-萘并萘醌目标化合物进行了基态构型优化,计算模型如图1所示。并通过振动分析确认了光异构化过程中所有光异构体的稳定态和过渡态。用Gaussian 09量子化学程序包完成全部计算。
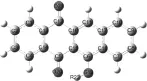
图1 萘并萘醌类化合物计算模型(R22:乙烯氧基—M11)
2 结果与讨论
2.1 光异构体结构
表1列出乙烯氧基萘并萘醌光异构化反应中所有光异构化化合物及其过渡态的几何构型数据。
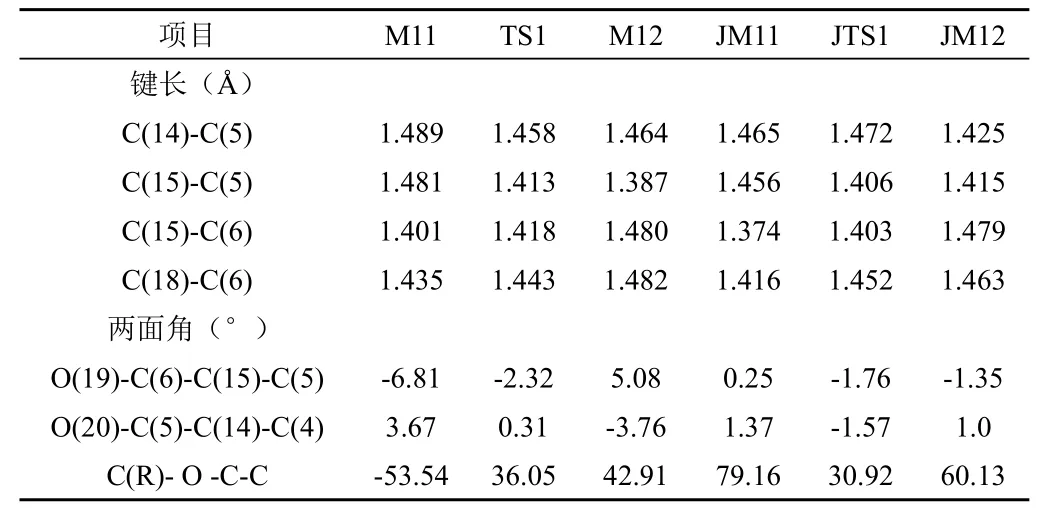
表1 6-乙烯氧基-5,12-萘并萘醌衍生物的光异构体的几何构型
表1的数据显示,在基态或激发态的光异构化过程中发生光异构化时,三种光异构化化合物的萘并萘醌结构中几个环之间呈现近共平面的变化规律。在过渡态时迁移基团与萘醌环平面形成的二面角为30°左右,基态和激发态时二面角的角度变大。这可能与迁移过程中因乙烯基基团体积小而有利于嵌入到O(19)原子和O(20)原子立体空间有关系。
2.2 原子净电荷分布
在B3LYP/6-31G水平上计算了6-乙烯氧基-5,12-萘并萘醌光异构体中各原子的Mulliken电荷分布列于表2。表2数据显示6种化合物负电荷集中在O(19),O(20),O(21)在原子上。

表2 6-乙烯氧基-5,12-萘并萘醌衍生物光异构体的自然原子电荷(e)
迁移基团乙烯基与母体醌环分子之间发生电荷转移,在基态以及激发态的构型中乙烯基均显示给电子性质,相互间电荷传递的净效果使得萘并萘醌分子带有-0.316e,-0.253e,-0.347 e,-0.392e,-0.37e,-0.449e的正电荷。乙烯基的给电子作用使得萘并萘醌环上各原子的负电荷基本上平均分布,最大负电荷都集中在了氧原子上。
2.3 光异构化反应
表3列出用密度泛函的B3LYP方法对乙烯氧基萘并萘醌化合物异构体进行构型优化之后,得到的能量数据,包括总能量,零点能,振动频率。
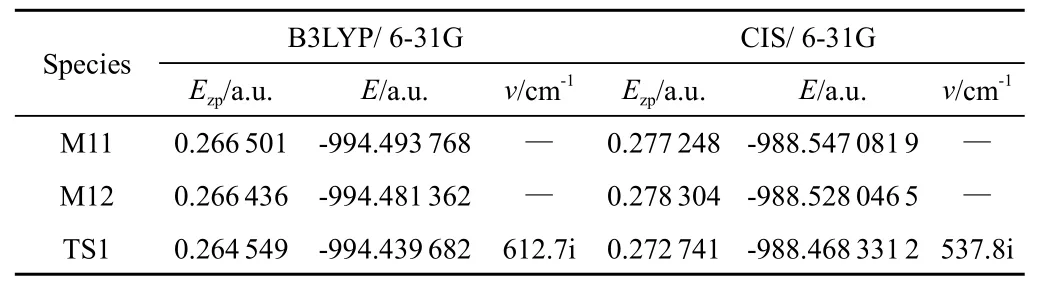
表3 6-乙烯氧基-5,12-萘并萘醌化合物的异构体和过渡态在基态和激发态下的零点能Ezp(a.u.),总能量E(a.u.)和谐振频率v(cm-1)
基态M11由过渡态TS1异构化为M12的反应的正活化能为147.25kJ/mol,逆活化能为111.32.76kJ/mol。JM11由过渡态JTS1异构化为JM12的反应的正活化能为161.9 kJ/mol,逆活化能为1 206.8kJ/mol。
迁移基团为乙烯基基团的萘并萘醌化合物与甲基基团[6]的化合物对比其光异构化反应活化能数据显示,正反应和逆反应活化能降低了40~90kJ/mol。迁移基团为乙烯基基团时其光异构化反应的活化能降低,说明不同迁移基团与萘并萘醌环上连接的O原子形成不同程度的共轭,以致对光异构反应的正逆活化能产生影响。
乙烯氧基萘并萘醌化合物的基态trans醌式构型M11比ana醌式构型M12构型能量低,M11稳定;激发态trans醌式构型JM11依然表现为比ana醌式构型JM12构型能量低。说明乙烯氧基萘并萘醌化合物能量上trans醌式构型有优势,以trans醌式构型存在。
2.4 紫外吸收光谱
对乙烯氧基萘并萘醌化合物光异构化反应中所有光异构体用TD/DFT进行了紫外吸收光谱计算,数据列于表4。光异构体的紫外光谱理论数据与实验测定值相符合,紫外光照射至达到光稳态,完成呈色过程:而后用可见光照射至光稳态,完成消色过程。
3 结论
6-乙烯氧基-5,12-萘并萘醌光致异构化反应中的活化能研究表明,在基态和激发态乙烯氧基萘并萘醌化合物两种光异构体都显示为trans醌式构型稳定,说明热力学上乙烯氧基萘并萘醌化合物有利于以trans醌式构型存在。乙烯氧基增加了共轭程度,可以有效地降低光异构化反应的活化能。对乙烯氧基萘并萘醌化合物光异构化反应中所有光异构体用TD/DFT进行了紫外吸收光谱计算,光异构体的紫外光谱理论数据与实验测定值相一致,与光异构化反应的光激发条件相符合。
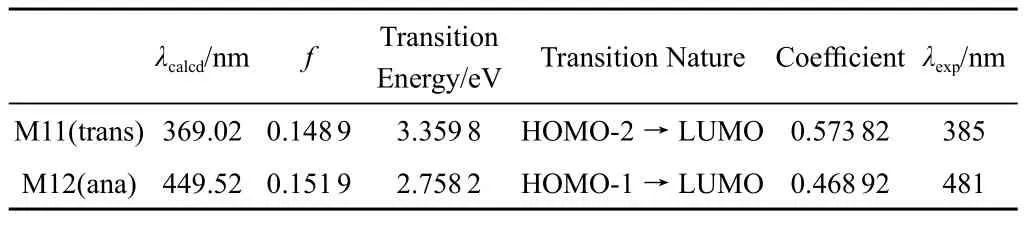
表4 M11在二甲亚砜溶液中的紫外光谱数据
[1] Irie M.Diarylethenes for Memories and Switches[J].Chemical Reviews,2000,100(5):1685-1716.
[2] Satapa山 y R,Padhy H,Wu Y.H,et a1.SyntIlesis and Characterization of Reversible Chemosensory Polymers:Modulation of Sensitivity through the Attachment of Novel ImidazolePendants[J]. Chemistry—A European Joumal,2012,18(50):1606l-16072.
[3] Lee S,Flood A H.Photoresponsive receptors for binding and releasing anions[J].Joumal of Physical Organic Chemistry,2013,26(2):79-86.
[4] Akiyama M.Blue—green light photochromism in europlum doped BaMgSiO4[J].Applied Physics Letters,2010,97:181903-18l905.
[5] Buchholtz,F.,Zelichenok,A.,Krongauz,V.Synthesis of new photochromic polymers based on phenoxynaphthacenequinone[J]. Macromolecules,1993,(26):906-910.
[6] 尹京花,连慧琴,吴学.6-羟基-5,12-萘并萘醌及其CH3,C6H5取代衍生物的光致变色理论研究[J].化学学报,2007,(65):2821-2826.
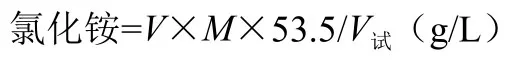
式中M—0.1mol/L硝酸银标准溶液的浓度;
V—见分析步骤消耗0.1mol/L硝酸银标准溶液的体积(mL);
参考文献
[1] 邹群,徐红娣.电镀溶液分析技术[M].化学工业出版社,2010.
Theoretical Study on Photochromic Properties of 6-Vinyl oxo-5,12-NaphthaLene Quinone
Yin Jing-hua
the CIS/6-31G method with B3LYP/6-31G method and ab initio density functional theory is studied respectively 6-ethyleneoxy-5,12-naphathacenequinone ground and excited states of the isomerization reaction.The reaction potential energy surface display,6-ethyleneoxy-5,12-naphathacenequinone in photoisomerization in ground and excited states cannot constitute the four level reaction.Thermodynamics is conducive to trans quinone configurations.In addition using the TD/B3LYP method in the presence of a solvent to calculate the UV absorption spectra of these compounds,the spectral data and the calculated values are basically the same,and photoisomerization of light excitation conditions are consistent.
naphthalene quinone;photochromism;photoisomerization;density functional theory
O641
B
1003-6490(2016)11-0059-02
2016-11-11
尹京花(1974—),女,吉林延吉人,副教授,主要研究方向为反应机理理论。
猜你喜欢
系统仿真技术(2022年4期)2023-01-17 13:01:44
食品工业(2022年11期)2022-11-28 03:26:48
数学物理学报(2022年3期)2022-05-25 13:33:22
食品与发酵工业(2022年5期)2022-03-30 09:01:08
数学物理学报(2022年1期)2022-03-16 06:15:04
云南化工(2021年8期)2021-12-21 06:37:38
数学物理学报(2021年5期)2021-11-19 07:01:16
数学物理学报(2021年3期)2021-07-19 06:02:18
广州化学(2020年6期)2020-12-28 06:53:12
黑龙江八一农垦大学学报(2020年2期)2020-05-06 03:34:48
