
琥珀酸索利那新片自研制剂与原研制剂溶出曲线的相似性评价
2016-11-19 03:27龚俊强徐彩虹吴春梅傅旭春浙江大学药学院杭州10058浙江华义医药有限公司浙江义乌000华东医药生物工程研究所杭州10011
中国药房 2016年30期
龚俊强,陈 仙,徐彩虹,张 霞,吴春梅,王 萍,傅旭春(1.浙江大学药学院,杭州 10058;.浙江华义医药有限公司,浙江义乌 000;.华东医药生物工程研究所,杭州 10011)
琥珀酸索利那新片自研制剂与原研制剂溶出曲线的相似性评价
龚俊强1,2*,陈 仙2,徐彩虹2,张 霞3,吴春梅2,王 萍2,傅旭春1#(1.浙江大学药学院,杭州 310058;2.浙江华义医药有限公司,浙江义乌 320002;3.华东医药生物工程研究所,杭州 310011)
目的:考察琥珀酸索利那新片自研制剂与原研制剂溶出曲线的相似性,为前者的处方和工艺筛选以及与后者的质量一致性评价提供参考。方法:采用桨法,转速为50 r/min,分别以水、pH1.2盐酸溶液、pH4.5乙酸盐缓冲液和pH6.8磷酸盐缓冲液为溶出介质进行溶出度试验,采用高效液相色谱法(HPLC)测定琥珀酸索利那新片自研制剂与原研制剂中主成分在不同时间点的累积溶出度,并绘制溶出度曲线,进而采用相似因子(f2)法评价二者溶出曲线的相似性。结果:在4种溶出介质中,琥珀酸索利那新片自研制剂与原研制剂的f2均>50,说明二者溶出曲线具有相似性。结论:所建立的HPLC法适用于琥珀酸索利那新片的溶出度测定;自研制剂与原研制剂的溶出行为基本一致,一定程度说明自研制剂的处方和工艺具有可行性。
琥珀酸索利那新片;自研制剂;原研制剂;高效液相色谱法;溶出曲线;相似因子法;评价
琥珀酸索利那新(Solifenacin succinate)是由日本安斯泰来制药公司开发,其片剂商品名为卫喜康(Vesicare),2004年8月首次在欧洲上市,同年11月获美国食品与药品管理局(FDA)许可在美国上市,2009年9月在我国获得上市许可。该药通过阻滞膀胱平滑肌的毒蕈碱M3受体来抑制逼尿肌的过度活动,从而缓解膀胱过度活动症(OAB)伴随的急迫性尿失禁、尿急和尿频症状。该药具有高度膀胱选择性和M3受体选择性,临床疗效确切,口干等副作用相对较少,患者耐受性好[1]。因此,包括中国、美国等多个国家的泌尿外科疾病诊断治疗指南都已将该药列为治疗OAB的一线药物[2-3]。
本研究中,笔者参考国家食品药品监督管理总局(CFDA)发布的《普通口服固体制剂溶出度试验技术指导原则》[4]和FDA溶出度方法数据库[5]等,对琥珀酸索利那新片自研制剂与原研制剂在水、pH1.2盐酸溶液、pH4.5乙酸盐缓冲液和pH6.8磷酸盐缓冲液4种溶出介质中溶出曲线的相似性进行了评价,旨在为其自研制剂的处方和工艺筛选以及与原研制剂的质量一致性评价提供参考。
1 材料
1.1 仪器
e2695型高效液相色谱仪,含自动进样器、光电二极管阵列检测器[沃特世科技(上海)有限公司];XS205DU型电子分析天平[梅特勒-托利多仪器(上海)有限公司];ZRS-8L型智能溶出试验仪(天津市天大天发科技有限公司)。
1.2 药品与试剂
琥珀酸索利那新对照品(埃斯特维华义制药有限公司,批号:13-601,纯度:99.9%);琥珀酸索利那新原料药(埃斯特维华义制药有限公司,批号:130401,纯度:99.2%);琥珀酸索利那新片原研制剂[商品名:卫喜康,安斯泰来制药(中国)有限公司,批号:12I22/03,规格:5 mg];琥珀酸索利那新片自研制剂(浙江华义医药有限公司,批号:130901,规格:5 mg);盐酸、氢氧化钠、乙酸、乙酸钠、磷酸氢二钾、磷酸二氢钾、磷酸(均为分析纯,国药集团化学试剂有限公司);乙腈(色谱纯,默克股份两合公司);试验用水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:Symmetry Shield RP18(150 mm×4.6 mm,5 μm);流动相:0.05 mol/L磷酸氢二钾缓冲液(pH6.0)-乙腈(65∶35,V/V);流速:1 ml/min;柱温:40℃;检测波长:210 nm;进样量:50 μl。
2.2 溶液的制备
2.2.1 对照品溶液 取琥珀酸索利那新对照品约25 mg,精密称定,置于50 ml量瓶中,以水-乙腈(70∶30,V/V)溶解并稀释至刻度,摇匀,得质量浓度为0.5 mg/ml的对照品贮备液。精密量取上述贮备液1.0 ml,置于100 ml量瓶中,用水-乙腈(70∶30,V/V)稀释至刻度,制成质量浓度为5 μg/ml的对照品溶液。
2.2.2 供试品溶液 取自研制剂样品,按2015年版《中国药典》(四部)“溶出度与释放度测定法”第二法(桨法)[6],以900 ml水为溶出介质,转速为50 r/min,温度(37.0±0.5)℃,依法操作,经30 min后,取溶出液适量,滤过,取续滤液3.5 ml与1.5 ml乙腈混匀,作为供试品溶液。
2.2.3 阴性对照溶液 按自研制剂处方取空白辅料适量,按“2.2.2”项下方法进行操作,制成阴性对照溶液。
2.2.4 溶出介质的制备 水:纯化水;pH1.2盐酸溶液:取盐酸7.65 ml,加水稀释至1 000 ml,摇匀,即得;pH4.5乙酸盐缓冲液:取2 mol/L乙酸溶液(取120.0 g冰乙酸用水稀释至1 000 ml,即得)14.0 ml和乙酸钠2.99 g,加水溶解并稀释至1 000 ml,摇匀,即得;pH6.8磷酸盐缓冲液:取0.2 mol/L磷酸二氢钾溶液(取27.22 g磷酸二氢钾,用水溶解并稀释至1 000 ml,即得)250 ml和0.2 mol/L氢氧化钠溶液(取8.00 g氢氧化钠,用水溶解并稀释至1 000 ml,即得)112.0 ml,加水稀释至1 000 ml,摇匀,即得。
2.3 专属性试验
取“2.2”项下对照品溶液、供试品溶液、阴性对照溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。结果表明,对照品与供试品的主峰保留时间一致,阴性对照在此波长下无干扰。

图1 高效液相色谱图A.对照品溶液;B.供试品溶液;C.阴性对照溶液;1.琥珀酸索利那新Fig 1 HPLC chromatogramsA.reference substance solution;B.test sample solution;C.negative control solution;1.solifenacin succinate
2.4 线性关系考察
取“2.2.1”项下对照品贮备液适量,用水-乙腈(70∶30,V/V)稀释成质量浓度约为0.4、0.8、2.0、3.2、4.0、4.8和5.6 μg/ml的系列对照品溶液,按“2.1”项下色谱条件进样测定。以待测成分质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程为y=133 495x-6 840(r=0.999 9)。结果表明,琥珀酸索利那新检测质量浓度线性范围为0.4~5.6 μg/ml。
2.5 精密度试验
取“2.4”项下0.4、4.0、5.6 μg/ml 3种质量浓度的琥珀酸索利那新对照品溶液适量,分别按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,琥珀酸索利那新峰面积的RSD分别为0.88%、0.20%、0.17%(n=6),表明仪器精密度良好。
2.6 稳定性试验
取自研制剂样品,以水、pH1.2盐酸溶液、pH4.5乙酸盐缓冲液和pH6.8磷酸盐缓冲液4种溶出介质,分别按“2.2.2”项下方法进行溶出度试验并制备供试品溶液,分别于室温下放置0、2、4、6、12 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,4种溶出介质中琥珀酸索利那新峰面积的RSD分别为0.45%、0.68%、0.70%和1.08%(n=5),表明供试品溶液在室温下放置12 h内稳定性良好。
2.7 重复性试验
取自研制剂样品,每次6片,按“2.2.2”项下方法进行溶出度试验并制备供试品溶液,再按“2.1”项下色谱条件进样测定并计算其溶出度,重复测定6次。结果,琥珀酸索利那新溶出度的平均值为97.60%,RSD=0.89%(n=6),表明本方法重复性良好。
2.8 回收率试验
根据自研制剂处方,按处方量50%、80%、100%精密称取琥珀酸索利那新对照品,辅料按处方量100%称取,每个水平各3份,按“2.2.2”项下方法进行溶出度试验并制备供试品溶液;另取琥珀酸索利那新对照品适量,精密称定,以水-乙腈(70∶30,V/V)溶解并稀释制成与供试品溶液质量浓度相近的对照品溶液。分别取上述两种溶液各适量,按“2.1”项下色谱条件进样测定,记录峰面积,以外标法计算回收率,结果见表1。
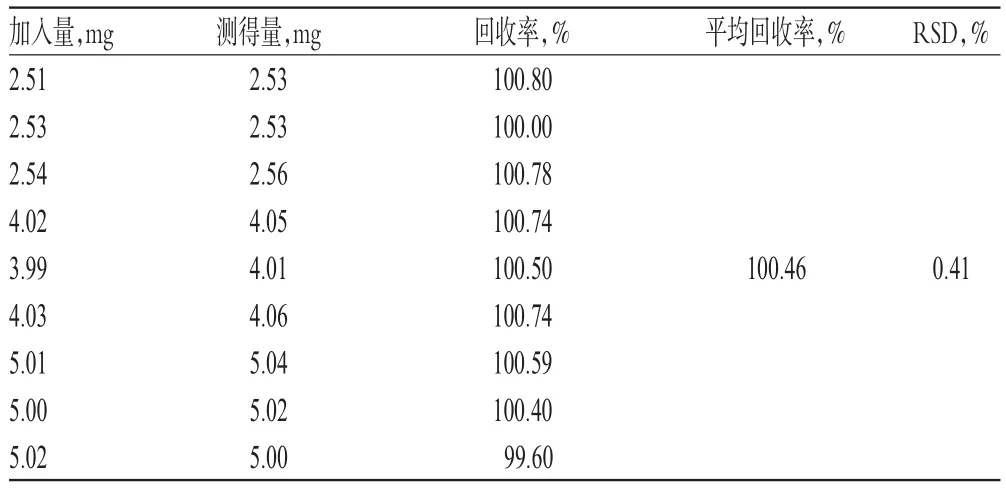
表1 回收率试验结果(n=9)Tab 1 Results of recovery tests(n=9)
2.9 溶出度测定及溶出曲线绘制
按2015年版《中国药典》(四部)“溶出度与释放度测定法”第二法(桨法)进行溶出试验[6],分别以900 ml水、pH1.2盐酸溶液、pH4.5乙酸盐缓冲液和pH6.8磷酸盐缓冲液作为溶出介质,转速50 r/min,温度(37.0±0.5)℃,依法操作,分别于5、10、15、20、30、45 min时取溶出液10 ml(即时在溶出杯中补加10 ml溶出介质),经0.45 μm微孔滤膜滤过后,取续滤液3.5 ml与1.5 ml乙腈混匀,制成供试品溶液;另取琥珀酸索利那新对照品适量,按“2.2.1”项下方法制成对照品溶液。分别取上述两种溶液各适量,按“2.1”项下色谱条件进样测定,记录峰面积,以外标法计算各时间点的累积溶出度,绘制溶出曲线,结果见表2和图2。

表2 琥珀酸索利那新片自研制剂与原研制剂在各溶出介质中的平均累积溶出度(n=12,%)Tab 2 Average cumulative dissolution of self-development and original preparations of Solifenacin succinate tablet in each dissolution medium(n=12,%)

图2 琥珀酸索利那新片自研制剂与原研制剂在各溶出介质中的溶出曲线(n=12)A.水;B.pH1.2盐酸溶液;C.pH4.5乙酸盐缓冲液;D.pH6.8磷酸盐缓冲液Fig 2 The cumulative dissolution profiles of self-development and original preparations of Solifenacin succinate tablet in each dissolution medium(n=12)A.water;B.pH1.2 hydrochloric acid solution;C.pH4.0 acetate buffer solution;D.pH6.8 phosphate buffered solution
2.10 溶出曲线的相似性评价
采用FDA推荐的相似因子(f2)法,分别考察在4种溶出介质中琥珀酸索利那新片自研制剂与原研制剂溶出曲线的相似性。f2计算公式为:

式中,Rt为原研制剂(参比制剂)t时间的平均累积溶出度;Tt为自研制剂(受试制剂)t时间的平均累积溶出度;n为取样时间点个数(n=3~5,且Rt>85%的点不超过1个,因此本研究选取5、10、15、30 min时的平均累积溶出度进行计算)。f2取值在0~100之间,FDA规定当f2值在50~100之间时,表明两种制剂的溶出曲线相似,且值越大相似度越高。经计算,琥珀酸索利那新片自研制剂与原研制剂在4种溶出介质中溶出曲线的f2均>50,说明二者溶出曲线具有相似性。琥珀酸索利那新片自研制剂与原研制剂在各溶出介质中溶出曲线的见表3。

表3 琥珀酸索利那新片自研制剂与原研制剂在各溶出介质中溶出曲线的f2(n=4)Tab 3 The f2of dissolution profiles of self-development and original preparations of Solifenacin succinate tablet in each dissolution medium(n=4)
3 讨论
体外溶出度试验常用于指导药物制剂的研发,评价制剂批间、批内质量的一致性,以及评价药品处方工艺变更前后质量和疗效的一致性等。已有数据表明,若仿制制剂体外多条溶出曲线与原研制剂皆一致,则二者体内生物利用度一致的概率高[7]。琥珀酸索利那新血药峰浓度(cmax)在口服后3~8 h达到[8],该药体内生理pH介质下30 min已经完全溶出,故其体外溶出曲线更重要的意义在于为制剂处方和工艺的开发及优化提供参考。尤其对于小规格口服固体制剂而言,原辅料的混合均匀性、片重和硬度差异等均会对溶出曲线和片间溶出差异产生较大影响。
3.1 溶出介质的选择
固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透。溶出度试验应尽可能在生理条件下进行,这样可以从药品体内行为的角度,更好地理解体外溶出数据[9],且溶出介质中药物溶解性应满足漏槽条件的要求。预试验中,取琥珀酸索利那新原料药约1 g,分别加入1 ml水、pH1.2盐酸溶液、pH4.5乙酸盐缓冲液和pH6.8磷酸盐缓冲液中,37℃恒温,均迅速溶解,呈无色透明黏稠状溶液,表明琥珀酸索利那新在所选取的不同pH介质中均易溶,溶解性为非pH依赖型,完全满足漏槽条件。故参考CFDA发布的《普通口服固体制剂溶出度试验技术指导原则》,最终选择水、pH1.2盐酸溶液、pH4.5乙酸盐缓冲液和pH6.8磷酸盐缓冲液作为溶出介质。
3.2 溶出方法和转速的选择
3.2.1 溶出方法的选择 取原研制剂(12片),按2015年版《中国药典》(四部)“溶出度与释放度测定法”项下第一法(篮法)与第二法(桨法)进行溶出试验[6],以水为溶出介质,溶出介质体积为900 ml,转速为50 r/min,分别在5、10、15、20、30、45 min时取溶出液10 ml(即时在溶出杯中补加10 ml溶出介质),经0.45 μm微孔滤膜滤过,取续滤液3.5 ml与1.5 ml乙腈混匀,按“2.1”项下色谱条件进样测定,记录峰面积并计算各取样时间点琥珀酸索利那新的累积溶出度。结果表明,采用篮法与桨法在各取样时间点的平均累积溶出度基本一致,因片剂溶出度测定一般采用桨法,故本试验最终选用桨法。
3.2.2 转速的选择 取原研制剂(12片),按2015年版《中国药典》(四部)“溶出度与释放度测定法”项下第二法(桨法)[6],以水为溶出介质,溶出介质体积为900 ml,转速分别为50、75 r/min,余按“3.2.1”项下方法操作,进样测定并计算各取样时间点琥珀酸索利那新的累积溶出度。结果表明,转速为75 r/min时溶出偏快,15 min时间点平均累积溶出度>85%,导致溶出曲线的区分能力较差;而当转速为50 r/min时,溶出速率较为合适,故本试验最终选择转速为50 r/min。
3.3 本研究的成果
本研究建立了琥珀酸索利那新片溶出度测定的HPLC法。经验证,本方法准确性好、灵敏度高,适用于琥珀酸索利那新片的溶出度测定。同时,本研究建立了具有区分力的琥珀酸索利那新片的溶出试验方法[10],为琥珀酸索利那新片自研制剂的处方和工艺筛选以及与原研制剂的质量一致性评价提供了试验依据。研究结果表明,自研制剂与原研制剂溶出行为基本一致,一定程度说明自研制剂的处方和工艺具有可行性,但二者的体内生物利用度和生物等效性是否一致还有待进一步考察。
[1] 郑建洪,黄红萍.治疗膀胱过度活动症的新药索利那新[J].中国新药与临床杂志,2013,32(10):771.
[2]那彦群,叶章群,孙颖浩,等.中国泌尿外科疾病诊断治疗指南:2014版[M].北京:人民卫生出版社,2013:330-333.
[3] Apostolidis A.Antimuscarinics in the treatment of OAB:is there a first-line and a second-line choice?[J].Current Drug Targets,2015,16(11):1 187.
[4]国家食品药品监督管理总局.普通口服固体制剂溶出度试验技术指导原则[EB/OL].(2015-02-05)[2015-06-06]. http://www.sda.gov.cn/WS01/CL0087/114286.html.
[5]FDA.Dissolution Methods Database:Solifenacin Succinate Tablets[EB/OL].(2008-02-19)[2015-07-06].http:// www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm.
[6]国家药典委员会.中华人民共和国药典:四部[S].2015年版.北京:中国医药科技出版社,2015:121-124.
[7]林兰,牛剑钊,许明哲,等.国外仿制药一致性评价比较分析[J].中国新药杂志,2013,22(21):2 470.
[8]刘军,姚建.新型毒蕈碱受体拮抗剂solifenacin治疗膀胱过动症[J].世界临床药物,2006,27(1):39.
[9]张启明,谢沐风,宁保明,等.采用多条溶出曲线评价口服固体制剂的内在质量[J].中国医药工业杂志,2009,40(12):946.
[10]谢沐风.具有区分力的溶出曲线[J].中国医药工业杂志,2014,45(7):687.
(编辑:周 箐)
Similarity Evaluation of Dissolution Profiles of Self-development and Original Preparation of Solifenacin Succinate Tablet
GONG Junqiang1,2,CHEN Xian2,XU Caihong2,ZHANG Xia3,WU Chunmei2,WANG Ping2,FU Xuchun1(1.College of Pharmacy,Zhejiang University,Hangzhou 310058,China;2.Zhejiang Huayi Pharmaceutical Co.,Ltd.,Zhejiang Yiwu 320002,China;3.Huadong Biological Engineering Research Institute,Hangzhou 310011,China)
OBJECTIVE:To explore the similarity of dissolution profiles of self-development and original preparation of Solifenacin succinate tablet,and provide reference for the prescription and process screening of the former one and the quality similarity evaluation of the latter one.METHODS:The paddle method was adopted with rotational speed of 50 r/min,using water,pH1.2 hydrochloric acid solution,pH4.0 acetate buffer solution and pH6.8 phosphate buffer solution as dissolution media,HPLC was used to determine the cumulative dissolution of main components of self-development and original preparation of Solifenacin succinate tablet at different time points,dissolution profile was drew,then f2was used to evaluate its similarity.RESULTS:In the 4 dissolution media,the f2of both self-development and original preparation of Solifenacin succinate tablet was higher than 50,which indicated that the dissolution profiles showed similarity.CONCLUSIONS:The established HPLC is suitable for the dissolution determination of Solifenacin succinate tablet;the dissolution profiles of the self-development and original preparations are basically similar,which indicates the prescription and technology of self-development preparation are feasible.
Solifenacin succinate tablet;Self-development;Original preparation;HPLC;Dissolution profiles;f2similarity factor;Evaluation
R927
A
1001-0408(2016)30-4311-04
2015-11-04
2016-09-14)
*工程师,硕士研究生。研究方向:药物制剂研发。E-mail:47160688@qq.com
#通信作者:教授,博士。研究方向:新药研发和药动学。E-mail:fuxc@zucc.edu.cn
DOI 10.6039/j.issn.1001-0408.2016.30.44
猜你喜欢
中国科技纵横(2021年24期)2021-03-02
蚕桑通报(2020年1期)2020-07-10
中国土壤与肥料(2018年5期)2018-11-05
中国酿造(2018年9期)2018-11-05
中成药(2018年6期)2018-07-11
中成药(2018年1期)2018-02-02
浙江工业大学学报(2017年5期)2018-01-22
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
中成药(2017年6期)2017-06-13
