
BSA与羟磷灰石相互吸附的FTIR-ATR光谱
2016-11-08 06:00周剑章叶艺文许朝曦林昌健林种玉厦门大学化学化工学院化学系固体表面物理化学国家重点实验室福建厦门6005浙江师范大学初阳学院浙江金华004厦门阿尔特系统工程有限公司福建厦门6005
物理化学学报 2016年2期
叶 青 胡 仁 周剑章 叶艺文 许朝曦 林昌健 林种玉,*(厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门6005;浙江师范大学初阳学院,浙江金华004;厦门阿尔特系统工程有限公司,福建厦门6005)
BSA与羟磷灰石相互吸附的FTIR-ATR光谱
叶青2胡仁1周剑章1叶艺文1许朝曦3林昌健1林种玉1,*
(1厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门361005;2浙江师范大学初阳学院,浙江金华321004;3厦门阿尔特系统工程有限公司,福建厦门361005)
采用傅里叶变换红外衰减全反射(FTIR-ATR)光谱法对牛血清白蛋白(BSA)在羟磷灰石(HA) [Ca10(OH)2(PO4)6]表面不同时间的相互吸附作用进行了表征。在BSA溶液作用下,羟磷灰石表面的Ca2+、PO43-和OH-离子初始的溶解和再沉淀使得BSA与HA相互作用层层叠加,在HA表面形成从表层到次表层分子都包含有吸附的BSA的覆盖层,从而加深两者之间的相互作用。经红外差谱法处理过的相关ATR数据表明,BSA与HA之间的相互作用是快速的,并随时间变化进一步加强;来自HA上PO43-的P=O基团对蛋白质肽键的酰胺II带(―CNH)、多肽链的甲基(―CH3)和亚甲基(―CH2)上氢的吸附作用要比P―O快速而且强烈。Ca2+在该吸附过程中起了极其重要的作用,其快速与蛋白质肽键的羰基氧发生作用,并诱导该蛋白质二级结构由β-折叠向α-螺旋和β-转角构象转变;伴随着这一构象变化,蛋白质多肽链上大多数肽键的―C=O和H―N―活性基团从链间氢键交联中释放出来,带动众多的氢分别参与同HA表面的Ca2+、PO43-和OH-离子的相互吸附作用,并牢牢地结合于HA表面;这对硬组织的再生起着重要作用,促进了HA的生物矿化过程。
生物材料;蛋白质吸附作用;多肽;红外光谱;FTIR-ATR;生物矿化作用
O646
doi:10.3866/PKU.WHXB201511301
1 引言
蛋白质的吸附作用有强吸附和弱吸附1,强吸附作用对各种生物过程产生着重要的影响,首先是对植入材料的生物反应的影响。相关作用的研究普遍侧重于测定各种生物材料表面吸附蛋白质的二级结构构象情况2-8。作为生物活性陶瓷的羟磷灰石与蛋白质之间的相互吸附作用过程也已有诸多的报道6-10,但相关过程的分子作用机制以及吸附的蛋白质是以何种分子构象为主乃存在争议,还有待进一步探讨。本文在前期工作10的基础上,采用傅里叶变换红外衰减全反射(FTIR-ATR)光谱法进一步考察了牛血清白蛋白在HA表面的吸附作用,揭示它们之间在各不同时间的相互作用机理。FTIR-ATR光谱法直接提供了吸附于HA表面的蛋白质的相关信息,但应该注意的一个重要问题是,由于ATR反射光透过深度(红外渐消场在酰胺I和酰胺II带区间的贯穿深度大约是400-600 nm)超过HA修饰层的厚度(约为20-40 nm),而进入BSA溶液中,该检测所得到的是表面吸附的和非吸附的蛋白质的混合光谱11。因此必须利用红外差谱方法2,11减去那部分非吸附的蛋白质的贡献,才能得到准确的吸附于HA表面的蛋白质信息。此方法提供了可信赖的而且重现性较好的差减结果12。另外,由于水在1643 cm-1处有较强的羟基弯曲振动吸收,在一定程度上干扰了蛋白质酰胺I带而影响观察其吸收情况。本实验采用重水(D2O)代替水溶液。
2 实验方法
2.1主要试剂
BSA(0903,99.0%)为上海生物工程有限公司生产的生化试剂。
2.2样品制备
锗晶体表面羟磷灰石的修饰按照文献9,10报道的方法进行。将几何尺寸为50 mm×10 mm×3 mm的ATR锗晶体侧立浸泡于沸腾的饱和Ca(OH)2溶液40 min,再用20%的H3PO4溶液(pH 2.0-2.4)于90°C继续浸泡30 min,之后将该晶体用去离子水冲洗2次在室温下晾干。牛血清白蛋白用0.9% NaCl重水溶液配制成浓度为40 mg∙mL-1的BSA溶液。
2.3FTIR-ATR检测
使用美国Nicolet 740 SX FT-IR光谱仪,MCTB检测器,衰减全反射附件。反射光束由锗晶体45°斜面入射,在晶体内产生多次的衰减全反射再经聚焦反射到达检测器检测。先测定锗晶体表面空白HA;之后往液池(带有HA修饰层的锗晶)中注入1 mL新鲜的BSA溶液,从0 min开始每隔1 min扫描收集数据至10 min,然后至15、30、60 min以及5、24和30h。扫描波长范围:4000-625cm-1,分辨率为4 cm-1,增益为1,扫描次数为128。为了检验测定结果的重现性,确保检测质量,该实验重复做3次。实验所测得的数据是HA表面吸附的和非吸附的BSA的混合光谱。为了获得准确的吸附于HA表面的BSA的信息,利用红外差谱法,调节各个差减因子(FCR)扣除那部分非吸附的蛋白质的吸收。
3 结果与讨论
3.1羟磷灰石及其对BSA的吸附作用
3.1.1锗晶体表面羟磷灰石修饰层
图1曲线1示出修饰于锗晶体表面羟磷灰石[Ca10(OH)2(PO4)6]的FTIR-ATR光谱图;与HA透射谱带101100-1000 cm-1区间出现的各吸收带频率相比较基本上接近,说明锗晶体表面的修饰层的确为HA,这样该修饰层含有Ca2+、PO34-和OH-羟磷灰石离子。其中1098和1026 cm-1吸收带均与HA的PO基团振动有关13,可分别归属于P=O和P―O的伸缩振动带。1656 cm-1的吸收为HA分子的O―H基团的弯曲振动带(该吸收带弱,并不影响观察蛋白质酰胺I带的吸收情况)。
图1曲线2和3分别为HA与BSA作用1 min和30 h的FTIR-ATR光谱图。谱线2和3上的蛋白质酰胺I带(1652,1649 cm-1)、酰胺II带(1548,1563 cm-1)以及蛋白质多肽链上的亚甲基(1459和1455 cm-1)和甲基(1445和1443 cm-1)的C―H弯曲振动带13-15的吸收实际上均为HA表面吸附的和非吸附的BSA的混合吸收光谱。作用1 min的P=O谱带由空白时的1098 cm-1红移至1094 cm-1,但作用了30 h该谱带还是在1094 cm-1处;而空白HA的P―O基团及其与BSA分别作用1 min和30 h的吸收均在1026 cm-1处。说明曲线2和3所示的1094与1026 cm-1的吸收应该分别来自HA分子中参与吸附和未参与吸附蛋白质的P=O与P―O各基团的混合吸收带。很可能由于参与吸附作用的PO(P=O与P―O)各基团含量较少,分别被还未参与吸附的各PO基团(含量多)的吸收所掩盖,因而看不出谱带的红移。1208 cm-1处的吸收归属于重水O―D的弯曲振动带。
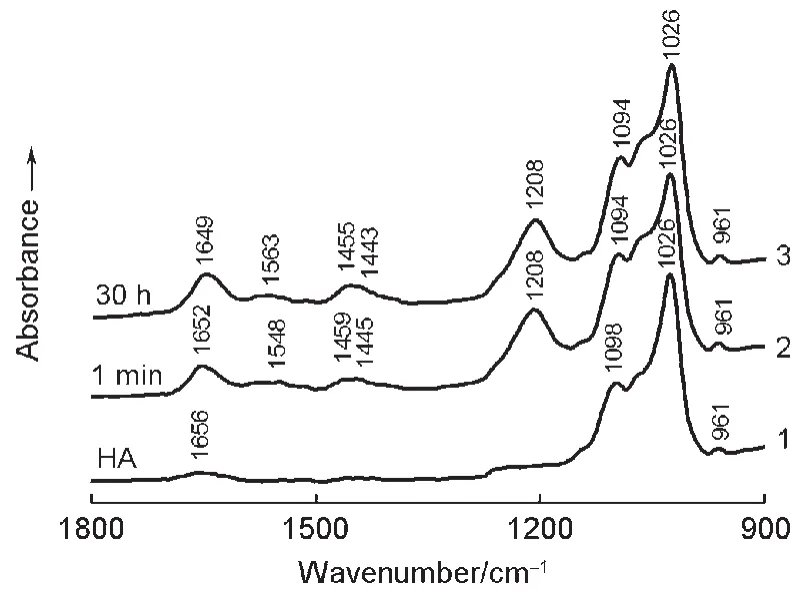
图1 锗晶体表面HA修饰层及其与BSA作用不同时间的FTIR-ATR光谱Fig.1 FTIR-ATR spectra of HAcoated on Ge crystal and reacted with BSAfor different time
3.1.2羟磷灰石修饰层的溶解与再沉淀
图2示出修饰于锗晶体表面的空白HA(曲线1)及其分别与BSA相互作用5 min,0.5 h,24 h和30 h的FTIR-ATR光谱图。由曲线2可以看出:加入BSA溶液的最初5 min,HA的P―O基团吸收带比曲线1明显减弱。说明加入BSA(NaCl/D2O)溶液后,HA修饰层与BSA发生了相互吸附作用;与此同时,BSA溶液中的Na+和Cl-很可能与涂层表面羟磷灰石离子进行离子交换6,16使部分Ca2+、与OH-溶解进入BSA溶液中;溶液中的蛋白质很容易吸附(结合)溶解出的PO34-基团,因此促使PO34-基团的进一步溶解(注意:这里PO34-基团进入溶液中的位置应该超出ATR反射光的透过深度),因此ATR检测到的各PO基团的吸收强度减弱。基团含有3个P―O键和1个P=O键(空白HA的P―O吸收带明显比P=O强),所以在最初5 min,随着PO34-基团的溶解,1026 cm-1处的P―O基团吸收带强度减弱要比P=O明显得多。文献6报道在初始的相同时间段里,含有白蛋白的盐溶液从磷酸钙涂层释放出的PO34-基团比空白盐溶液所释放的要多得多(前者约为后者的2.5倍多)。显然,蛋白质的吸附作用是影响HA修饰层溶解的重要因素6,17。
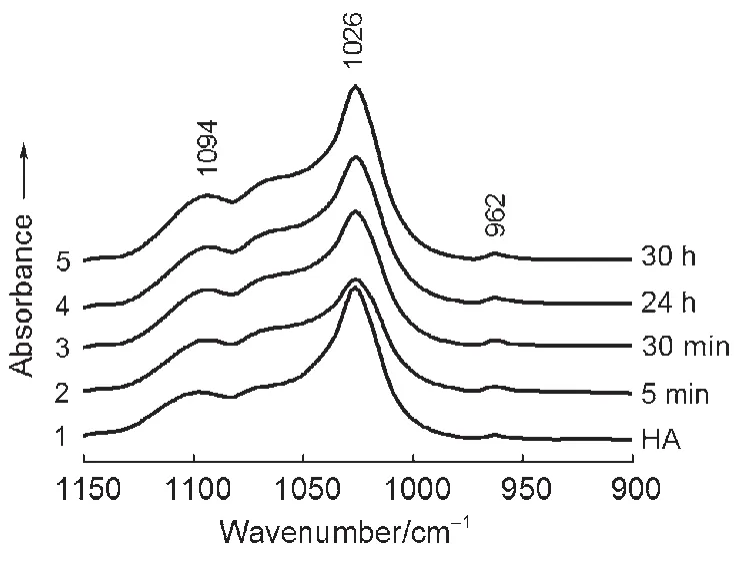
图2 HA修饰层及其与BSA作用不同时间的FTIR-ATR光谱Fig.2 FTIR-ATR spectra of HAcoating and reacted with BSAfor different time
随后吸附于HA涂层表面的生物大分子继续吸附溶液中的HA离子,使溶液中的Ca2+、和OH-离子快速沉淀下来。于是涂层上PO各基团吸收带逐渐增强(图2曲线3-5)。这部分被吸附而覆盖于蛋白质表面的HA离子紧接着又与溶液中的BSA发生吸附作用。HA表面的Ca2+、PO34-和OH-离子的这种初始的溶解和再沉淀9,18,19使得BSA与HA相互作用层层叠加,在HA修饰层形成从表层到次表层分子都包含有吸附的BSA覆盖层9,从而加深两者之间的相互作用。
3.1.3羟磷灰石(PO43-基团)对BSA的吸附作用
为了得到准确的参与吸附蛋白质的PO各基团的信息,利用IR差谱法扣除那部分还未参与吸附蛋白质的PO各基团的吸收。图3曲线1是与BSA作用1 min的HA扣除空白HA谱线的差减结果。该曲线较为准确地展示出作用1 min时参与吸附蛋白质的PO各基团的信息。谱线上HA的PO各基团由于参与吸附蛋白质,P=O基团的吸收由原来的1098 cm-1红移至1091和1064 cm-1,而P―O由1026红移至1015 cm-1。仅仅作用1 min时间,PO各吸收带就出现明显红移,充分表明HA对BSA的作用是快速的化学吸附。图3曲线2-5分别是与BSA作用30 min、60 min、5 h和30 h的HA减去空白HA的红外光谱差谱。各曲线分别清楚地展示出参与吸附蛋白质的P=O基团的红移:由1090和1063 cm-1(30 min)分别至1089和1063 cm-1(60 min、5 h和30 h)以及P―O基团由1013 cm-1(30 min)→1010 cm-1(60 min)→1009 cm-1(5 h)→1007 cm-1(30 h)。而且这些吸收带强度随时间变化明显增强;1063和1089 cm-1谱带增强尤为明显,表明P=O基团与蛋白质的相互吸附作用要比P―O基团快速而且强烈。PO各基团很可能是与蛋白质多肽的―CH3、―CH2和肽键的―C―N―H基团的氢发生吸附作用。从PO的价键结构特征分析:P―O单键为σ键而P=O双键除含有σ键外还含有π键。由于π键在化学性能上比σ键更活泼,更容易接受―CH和―CNH上的氢原子,因此含有π键的P=O基团与氢的相互作用要比P―O更加迅速而强烈。这与红外光谱检测分析结果完全一致。P=O基团对蛋白质的作用有强吸附(1063 cm-1)和弱吸附(1089 cm-1)两种情况,其强吸附作用比弱吸附要快速得多。
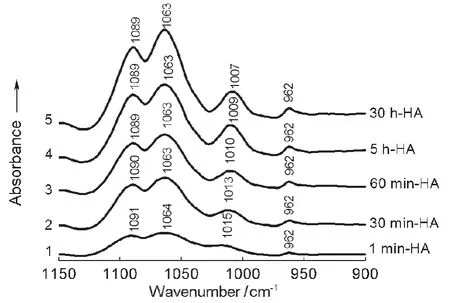
图3 与BSA作用不同时间的HA的FTIR-ATR光谱差谱Fig.3 FTIR-ATR subtraction results of HA reacted with BSAfor different time
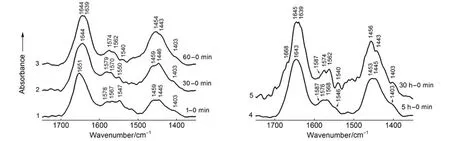
图4 与HA作用不同时间的BSA的FTIR-ATR光谱差谱Fig.4 FTIR-ATR difference spectra of BSAreacted with HAfor different time
3.2牛血清白蛋白在HA上的吸附作用
图4曲线1-5分别为与HA作用1 min、30 min、60 min、5 h和30 h的BSA减去与HA作用0 min的BSA光谱差谱。这里0 min谱线是经过128次扫描,历时64 s获得的,该谱线是HA表面吸附和非吸附的BSA混合光谱,但非吸附的占据大多数。减去该谱线时,虽然减去了一小部分初始吸附的BSA,但非吸附的BSA却全被扣除。而各谱线都同样扣除那一小部分初始吸附的BSA,并不影响各吸附的BSA谱线之间的对比分析结果。由于扣除了非吸附的BSA吸收的干扰,曲线1展示出与HA作用1 min后吸附于HA表面的蛋白质吸收带。1651 cm-1的吸收归属于肽键的酰胺I带C=O伸缩振动,说明该蛋白质的二级结构是以α-螺旋构象(这是蛋白质中含量最丰富的二级结构,体系中该谱带的中心位置应于1653 cm-1附近)成分为主3,20,21,应该还包含有β-折叠构象(这是蛋白质中第二种最常见的二级结构)成分所对应的振动吸收子带;该谱带的中心大约位于1632 cm-1附近,在此与α-螺旋构象所对应的振动吸收子带重叠在一起2,3,12,21。1459和1445 cm-1的吸收分别为多肽的―CH2和―CH3的C―H弯曲振动带。曲线1-3示出在重水中(1-60 min)谱带1540-1579 cm-1的吸收均处在肽键酰胺II带的范围内13-15,21(酰胺II带在酰胺质子氢-氘交换后,产生大的红移,移至1450 cm-1附近),说明由于该酰胺质子处于内埋或强的氢键作用而不容易交换,使氢-氘交换作用不完全21。
值得注意的是,图4曲线2示出BSA与HA作用30 min后,位于1579、1570和1550 cm-1的酰胺II带的吸收比作用1 min的各对应吸收带都有1-3 cm-1的蓝移。该谱带来自酰胺II带的N―H弯曲振动和C―N伸缩振动强偶合产生的吸收带13,14,21,谱带的蓝移说明N―H和C―N的键力有所增强。文献21,22表明,当有金属离子存在时,归属于蛋白质二级结构β-折叠的酰胺II带的吸收发生4-6 cm-1的蓝移(酰胺I带有1 cm-1的位移);并进一步指出,由于金属离子的影响,使得蛋白质(尤其是1634 cm-1处的β-折叠构象)有很大一部分的氢键作用发生了变化。因此可以推断体系中吸附的BSA的β-折叠的多肽链间氢键交联已经发生了变化。这里肽键的―C―N―H基团中C―N和N―H键力增强,说明体系中没参与相邻链间肽键(―N―H和O=C―)的氢键交联作用的―N―H含量增加了(可以确定没参与氢键作用的O=C―含量也随之增加);因为β-折叠中所有的肽键都参与了多肽链间氢键的交联作用23,这就是说原来β-折叠中相邻肽链间的氢键在Ca2+的作用下发生了断裂,转而由各自肽链结构的―N―H和O=C―形成5→1或4→1(肽单元)链内氢键的α-螺旋或β-转角23等的构象。由于Ca2+极易与蛋白质结合并诱导该蛋白质二级结构向α-螺旋转变24-29;又由于α-螺旋构象的规则性引起该螺旋折叠中的协同作用,一旦体系中形成链内第一圈α-螺旋,随后逐个残基的加入(即螺圈的形成)就会变得更加容易而迅速23。因此曲线2酰胺II带的蓝移说明体系中有一部分蛋白质二级结构由β-折叠转变为α-螺旋。在这种情况下,由于没参与氢键交联的―C=O基团含量增加,酰胺I带理应蓝移;但曲线2的酰胺I带却由原来的1651 cm-1红移至1644 cm-1。文献30指出:来自肽链侧链带负电荷的羧基氧和肽键上不带电荷的羰基氧都能很好地结合于Ca2+上,因此酰胺I带的红移应该是Ca2+与α-螺旋中没参加氢键交联作用的羰基氧发生了吸附作用,并进一步形成配位键3,15,30,使得C=O键力削弱,酰胺I带红移2,15,31。EDS检测出空白HA表面的Ca/P比值为1.259,说明该修饰层表面Ca2+的含量较高,能与较多酰胺I带的氧发生作用。这里酰胺I带的频率位置主要取决于两方面的因素:一是当肽链由β-折叠转变为α-螺旋时,由于没参与氢键交联作用的羰基氧含量增多,C=O键力增强而引起谱带蓝移;二是Ca2+与羰基氧的络合作用削弱C=O键力,而导致该谱带红移。由于后者的作用更为强烈,两者叠加的结果是C=O键力削弱引起谱带红移。之前发表的论文,由于体系中Pt(IV)32以及Au(III)33的含量很少,因此金属离子对羰基氧的作用所引起C=O键力的削弱程度比起由于肽链构象变化而释放出相当大一部分的羰基氧所引起的C=O键力增强,前者的作用显得微不足道(基本上可以忽略);净结果自然是C=O键力增强,导致谱带蓝移。因此判断蛋白质二级结构构象时,在金属离子含量较多的情况下,应该考虑到其与羰基氧的作用对C=O键力的影响。曲线2的1644 cm-1的吸收应该是吸附于HA表面的BSA中已经参与和还未参与同Ca2+作用的α-螺旋的C=O混合吸收带。
图4曲线3示出与HA作用60 min的BSA酰胺I带有一部分由原来的1644 cm-1红移至1639 cm-1处,而且酰胺II带各吸收也比作用30 min时的有明显红移,说明此时BSA与HA之间的相互作用进一步增强。
图4曲线4示出与HA作用5 h的BSA的酰胺I带由1644 cm-1红移至1643 cm-1,其原来的1639 cm-1的吸收应该被包在1643 cm-1谱带中。酰胺II带比作用60 min的各有2-6 cm-1的蓝移,说明此时肽链β-折叠的链间氢键在Ca2+的作用下继续断裂而转变为α-螺旋构象(与曲线2的情况相似)。
作用30 h的BSA(图4曲线5)在1587 cm-1处的肩峰和1403 cm-1的吸收分别来自肽链侧链及其C端的离子化羧基()的反对称和对称伸缩振动吸收带13,15,34-36,该吸收比作用5 h的明显增强,说明随作用时间延长,吸附于HA表面的羧基阴离子的量进一步增加。Ca2+应该是通过强大的静电作用力将羧基阴离子吸引到HA表面,两者之间靠离子键维系15,21,34,35。羧基阴离子比酰胺I带更容易与Ca2+发生配位作用24,25,36,形成配位羧基后,其反对称伸缩振动谱带由原来的1587 cm-1蓝移至1605 cm-1附近34,36;由于含量少(该红外吸收大约只占酰胺I带的10%-30%35,37),被强的酰胺Ⅰ带所掩盖21。1668 cm-1处出现的新峰来自β-转角构象的羰基吸收20,21,38,这是由各自肽链上肽键的―C=O与H―N―基团之间形成4→1的分子内氢键的又一种环形构象23。此外还可清楚地看到归属于α-螺旋构象的酰胺I带分裂为1645和1639 cm-1两个吸收峰(随后将对此作详细分析);该吸收带整体的强度随时间变化明显增强,说明有更多α-螺旋的羰基氧参与了同Ca2+的相互作用。曲线5的酰胺II带(1574、1562和1540 cm-1)以及1-5各条曲线的CH2(1459-1453 cm-1)和CH3(1446-1443 cm-1)的C―H弯曲振动吸收带也都随时间延长而增强,CH2吸收带的增强尤为明显(曲线5,1456 cm-1);说明随着越来越多酰胺I带与Ca2+的作用,带动更多肽键和碳氢键上的氢参与了同HA表面的PO和OH基团的吸附作用,并且牢牢地结合于HA表面。在这一吸附过程中,酰胺II带始终保持在正常的范围内;酰胺质子的氘代作用难于在Ca2+负载的状态下进行,暗示Ca2与肽键的结合相当紧密和坚实。进一步表明体系中的BSA与HA具有很强的相互作用能力。
图5曲线1-7示出与HA作用30 h的BSA分别扣除与HA作用0、1、2、3、5、10、15 min的BSA的光谱差谱。这里主要观察从0、1、2、3、5、10、15 min至30 h各不同时间段酰胺I带的吸收情况。其中1668 cm-1(来自β-转角构象的羰基吸收,其含量明显比较少)和1639 cm-1(主要来自与Ca2+发生作用的α-螺旋的酰胺I带)两处的吸收频率并不随各不同的时间段而变化,吸收强度也没有明显的改变;而1649 cm-1的吸收频率和强度却随各不同的时间段而变化。此时(已经作用了30 h),体系中绝大多数的Ca2+都参加了与羧基阴离子和α-螺旋的羰基氧的配位作用,已经达到了化学平衡;而肽链的α-螺旋还在形成。Chittur20指出,溶液中蛋白质α-螺旋的吸收发生在较低的波数,大约在1650-1655 cm-1附近;而在重水中该吸收会降得更低。因此,1649 cm-1的吸收应该主要来自这部分还没与Ca2+发生络合作用的α-螺旋的C=O吸收带的贡献。从曲线1至7(时间段朝后靠),该吸收呈增长趋势并由1645 cm-1(曲线1-4)→1646 cm-1(曲线5)→1648 cm-1(曲线6)→1649 cm-1(曲线7)呈现蓝移;表明随着时间段朝后推移,体系中没与Ca2+发生作用的α-螺旋的―C=O含量逐渐增多。这为该体系吸附的蛋白质二级结构构象变化趋势是由β-折叠转变为α-螺旋(和一小部分β-转角)提供了进一步的证据。
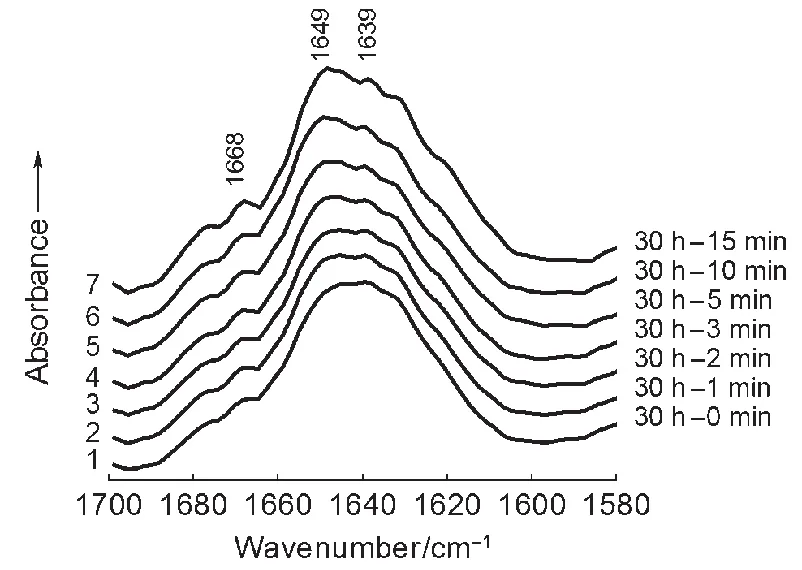
图5 系列与HA作用30 h的BSA的FTIR-ATR光谱差谱Fig.5 FTIR-ATR subtraction results of BSA reacted with HAfor 30 h
3.3α-螺旋构象促进蛋白质与HA之间的相互作用
Moulton等1观察到吸附于TiO2表面免疫球蛋白的酰胺I带(1636 cm-1)和酰胺II带(1545 cm-1)在加入磷酸盐缓冲液后两谱带吸收明显减弱,而且酰胺I带由1636 cm-1蓝移至1651 cm-1。这是由于原先吸附于TiO2表面的酰胺I带(1636 cm-1)是以β-折叠构象成分为主而其中还含有一部分α-螺旋构象成分所对应的振动吸收子带。当加入磷酸盐缓冲剂后,PO各基团参与了同TiO2表面该蛋白质酰胺I带的竞争吸附作用;取代了吸附于该表面酰胺I带中绝大部分的β-折叠(弱吸附)组分,导致1636 cm-1谱带消失只剩下α-螺旋组分所对应的吸收带(1651 cm-1)。充分说明α-螺旋构象的蛋白质具有很强的吸附能力,能牢牢地结合于TiO2生物材料表面。
β-折叠构象中所有肽键的―N―H和O=C―基团都参加了多肽链间的氢键交联作用;而α-螺旋和β-转角分别只有20%和25%的肽键参加链内的氢键交联。当BSA蛋白质二级结构构象由β-折叠转变为α-螺旋和β-转角时,其肽链上分别有相当多的―N―H和O=C―活性基团从链间氢键作用中释放出来参与同HA作用;而且随着肽链由伸展的折叠片向螺旋圈卷曲而引起该链及其侧链上各化学官能团的空间位置发生变化这一动态过程,带动更多的羧基阴离子、肽键以及大量的氢共同参与对HA的吸附作用,大大提高该蛋白质对HA的亲合性。另外,由于α-螺旋0.54 nm的螺距与Ca2+在HA六方晶系中(001晶面)的0.545 nm原子间点阵间距十分接近39,这一有意义的构象特征十分有利于蛋白质调节其自身与Ca2+的相互作用24,40,41。看来吸附的蛋白质的α-螺旋构象,其规律性间距的羰基氧和侧链羧基阴离子与处于这种点阵结构的Ca2+形成了高度的互补。这应该是蛋白质能牢固地吸附并结合于HA表面的重要原因。体系中BSA正是通过肽链α-螺旋表面大量的活性基团与HA之间的这种强的化学吸附作用来传递并调节细胞活性物质对HA界面的基本反应42,43,这对促进硬组织的再生起着十分重要的作用8,44-46。
4 结论
IR实验结果表明:吸附于HA表面的BSA在Ca2+的诱导作用下,其蛋白质二级结构由β-折叠转变为α-螺旋和一小部分β-转角的圈形构象,从而大大提高了蛋白质对HA生物材料的吸附活性,使蛋白质能牢牢地吸附并结合于HA表面。这样有利于细胞活性物质与HA界面的相互作用,从而引起和促进了生物矿化过程。
References
(1)Moulton,S.E.;Barisci,J.N.;McQuillan,A.J.;Wallace,G.G. Colloid.Surf.A:Physicochem.Eng.Asp.2003,220,159.doi: 10.1016/S0927-7757(03)00078-5
(2)Lenk,T.J.;Ratner,B.D.;Gendreau,R.M.;Chittur,K.K. J.Biomed.Mater.Res.1989,23,549.
(3)Shen,Y.H.;Yang,Z.L.;Wu,J.G.Acta Phys.-Chim.Sin. 1999,15(12),1064.[沈玉华,杨展澜,吴瑾光.物理化学学报,1999,15(12),1064.]doi:10.3866/PKU.WHXB19991203
(4)Shen,Y.H.;Yang,Z.L.;Wu,J.G.1999,35(4),431.[沈玉华,杨展澜,吴瑾光.北京大学学报(自然科学版),1999,35(4), 431.]
(5)Elangovan,S.;Margolis,H.C.;Oppenheim,F.G.;Beniash,E. Langmuir 2007,23(22),11200.doi:10.1021/la7013978
(6)Ong,J.L.;Chittur,K.K.;Lucas,L.C.J.Biomed.Mater.Res. 1994,28,1337.
(7)Yang,Q.;Zhang,Y.Y.;Liu,M.L.;Ye,M.;Zhang,Y.Q.;Yao, S.Z.Analytica Chimica Acta 2007,597,58.doi:10.1016/j. aca.2007.06.025
(8)Zeng,H.T.;Chittur,K.K.;Lacefield,W.R.Biomaterials 1999,20,377.doi:10.1016/S0142-9612(98)00184-7
(9)Feng,B.;Chen,J.Y.;Zhang,X.D.Biomaterials 2002,23 (12),2499.doi:10.1016/S0142-9612(01)00384-2
(10)Ye,Q.;Hu,R.;Lin,Z.Y.;Lin,C.J.Chem.J.Chin.Univ.2006, 27(8),1552.[叶青,胡仁,林种玉,林昌健.高等学校化学学报,2006,27(8),1552.]
(11)Yan,Y.L.;Jin,Z.C.Applications of Fourier Transform Infrared Spectrometry in Researches ofAgriculture,Food and Biology.In Modern Fourier-Transform Infrared Spectrometry and Its Applications(Part II);1st ed.;Wu,J.G.Ed.;Literature of Science and Technology Press:Beijing,1994;pp 177-180. [严衍禄,金泽宸.傅里叶变换光谱在农业、食品和生物学研究中的应用;近代傅里叶变换红外光谱技术及应用(下卷).吴瑾光主编.北京:科学技术文献出版社,1994:177-180.]
(12)Maruyama,T.;Katoh,S.;Nakajima,M.;Nabetani,H.;Abbott, T.P.;Shono,A.;Satoh,K.J.Membr.Sci.2001,192,201.doi: 10.1016/S0376-7388(01)00502-6
(13)Bellamy,L.J.The Infra-red Spectra of Complex Molecules, 3rd ed.;Chapman and Hall:London,1975;pp 14-26,107-125,183-202,386.
(14)Silverstein,R.M.;Bassler,G.C.;Morrill,T.C.Infrared spectrometry.In Spectrometric Identification of Organic Compounds,5th ed.;Sawicki,D.,Stiefel,J.Eds.;Wiley:New York,1991;pp 103,123.
(15)Rao,C.N.R.Chemical applications of Infrared Spectroscopy. 1st ed.;Academic Press Inc.:New York,1963;pp 131-138, 255-262,480-495.
(16)Driessens,F.C.M.;van Dijk,J.W.E.;Borggreven,J.M.P.M. Calcif.Tissue Res.1978,26,127.doi:10.1007/BF02013247
(17)Clark,G.C.F.;Williams,D.F.J.Biomed.Mater.Res.1982, 16(2),125.
(18)Ong,J.L.;Lucas,L.C.Biomaterials.1998,19(4-5), 455.doi:10.1016/S0142-9612(97)00224-X
(19)Margolis,H.C.;Moreno,E.C.Calcif.Tissue Int.1992,50(2), 137.doi:10.1007/BF00298791
(20)Chittur,K.K.Biomaterials 1998,19(4-5),357.doi:10.1016/ S0142-9612(97)00223-8
(21)Zhou,J.M.;Shi,N.Applications of Fourier Transform Infrared Spectrometry in Biochemistry.In Modern Fouriertransform Infrared Spectrometry and Its Applications(Part II), 1st ed.;Wu,J.G.Ed.;Literature of Science and Technology Press:Beijing,1994;pp 193-212.[周筠梅,施鼐.傅里叶变换红外光谱在生物化学中的应用;近代傅里叶变换红外光谱技术及应用(下卷).吴瑾光主编.北京:科学技术文献出版社,1994:193-212.]
(22)Alvarez,J.;Haris,P.I.;Lee,D.C.;Chapman,D.Biochimica Et Biophysica Acta 1987,916(1),5.doi:10.1016/0167-4838 (87)90204-4
(23)Xu,C.F.Proteins.In Biochemistry(Book One),2nd ed.;Shen, T.;Wang,J.Y.Eds.;Higher Education Press:Beijing,1990; pp 146-150.[徐长法.生物化学(上册).沈同,王镜岩主编.北京:高等教育出版社,1990:146-150.]
(24)Hauschka,P.V.Haemostasis 1986,16(3-4),258.
(25)Prestrelski,S.J.;Byler,D.M.;Thompson,M.P.Biochemistry 1991,30(36),8797.doi:10.1021/bi00100a010
(26)Fronticelli,C.;Bucci,E.;Shamoo,A.E.Biophys.Chem.1984, 19(3),255.doi:10.1016/0301-4622(84)87007-6
(27)Inesi,G.Annu.Rev.Physiol.1985,47,573.doi:10.1146/ annurev.ph.47.030185.003041
(28)Wu,G.R.;Gao,Z.Y.;Dong,A.C.;Yu,S.N.Int.J.Biol. Macromol.2012,50(4),1011.
(29)Zhao,Y.W.;Yang,H.Y.;Meng,K.J.;Yu,S.N.Int.J.Biol. Macromol.2014,64,453.doi:10.1016/j.ijbiomac.2013.12.036
(30)Yu,M.M.Hormones.In Biochemistry(Book One),2nd ed.; Shen,T.,Wang,J.Y.Eds.;Higher Education Press:Beijing, 1990;pp 447-455.[俞梅敏.生物化学(上册).沈同,王镜岩主编.北京:高等教育出版社,1990:447-455.]
(31)Morrissey,B.W.;Stromberg,R.R.J.Colloid.Interface Sci. 1974,46(1),152.doi:10.1016/0021-9797(74)90036-8
(32)Lin,Z.Y.;Xue,R.;Ye,Y.W.;Zheng,J.H.;Xu,Z.L.BMC Biotechnol.2009,9,62.doi:10.1186/1472-6750-9-62
(33)Lin,Z.Y.;Ye,Y.W.;Li,Q.L.;Xu,Z.L.;Wang,M.BMC Biotechnol.2011,11,98.doi:10.1186/1472-6750-11-98
(34)Zhou,W.J.;Wang,Y.Applications of Infrared Spectrometry in Basic Researches of Inorganic and Coordination Chemistry.In Modern Fourier-transform Infrared Spectrometry and Its Applications(Part II),1st ed.;Wu,J.G.Ed.;Literature of Science and Technology Press:Beijing,1994;pp 292-296.[周维金,王毅.红外光谱法在无机和配位化学基础研究中的应用;近代傅里叶变换红外光谱技术及应用(下卷).吴瑾光主编.北京:科学技术文献版社,1994:292-296.]
(35)Barth,A.Prog.Biophys.Mol.Biol.2000,74(3-5),141.doi: 10.1016/S0079-6107(00)00021-3
(36)Farkas,V.;Vass,E.;Hanssens,I.;Majer,Z.;Hollosi,M. Bioorg.Med.Chem.2005,13(17),5310.doi:10.1016/j. bmc.2005.06.040
(37)Rahmelow,K.;Hübner,W.;Ackermann,T.Anal.Biochem. 1998,257(1),1.doi:10.1006/abio.1997.2502
(38)Shai,Y.Biochim.Biophys.Acta-Biomembr.2013,1828(10), 2306.
(39)Kay,M.I.;Young,R.A.;Posner,A.S.Nature 1964,204, 1050.doi:10.1038/2041050a0
(40)Hauschka,P.V.;Carr,S.A.Biochemistry 1982,21(10), 2538.doi:10.1021/bi00539a038
(41)Hauschka,P.V.Osteocalcin and Its Functional Domains.In The Chemistry and Biology of Mineralized Tissues;Butler,W. T.Ed.;EBSCO Media:Birmingham,1985;pp 149-158.
(42)Horbett,T.A.ProteinAdsorption on Biomaterials.In Biomaterials:Interface Phenomena and Applications;Cooper, S.L.,Peppas,N.A.Eds.;American Chemical Society: Washington DC,1982;pp 233-244.
(43)Davies,J.E.The importance and Measurement of Surface Charge Species in Cell Behavior at the Biomaterial Interface. In Surface Characterization of Biomaterials;Ratner,B.D. Ed.;Elsevier:Amsterdam,1988;pp 219-222.
(44)Ono,I.;Gunji,H.;Kaneko,F.;Saito,T.;Kuboki,Y. J.Craniofac.Surg.1995,6(3),238.doi:10.1097/00001665-199505000-00011
(45)El-Ghannam,A.;Ducheyne,P.;Shapiro,I.M.Trans 21st Annual Meeting of Society for Biomaterials 1995,46.
(46)Gungormus,M.;Fong,H.;Kim,I.W.;Evans,J.S.;Tamerler, C.;Sarikaya,M.Biomacromolecules 2008,9(3),966.doi: 10.1021/bm701037x
FTIR-ATR Spectrometry of BSA Adsorption on Hydroxyapatite
YE Qing2HU Ren1ZHOU Jian-Zhang1YE Yi-Wen1XU Zhao-Xi3LIN Chang-Jian1LIN Zhong-Yu1,*
(1State Key Laboratory of Physical Chemistry of Solid Surfaces,Department of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,Fujian Province,P.R.China;2Chuyang Honors College,Zhejiang Normal University,Jinhua 321004,Zhejiang Province,P.R.China;3Xiamen AERTE System Engineering CO.,LTD,Xiamen 361005,Fujian Province,P.R.China)
The microcosmic process of bovine serum albumin(BSA)adsorbing onto hydroxyapatite(HA)for different time intervals was investigated by Fourier transform infrared attenuated total internal reflectance(FTIRATR)spectrometry.The initial dissolution and re-precipitation of PO43-,Ca2+,and OH-ions from the HAcoating led to the occurrence of the coating including adsorbed BSAon the HA from surface-to subsurface-molecular layers and to in-depth interaction between BSAand HA.The subtraction results gained in the adsorption regions of HAand BSAreveal that the binding of P=O,from the phosphate(PO43-),tothehydrogenof amide II,methyl and methene of the BSA appears to be considerably more rapid and stronger than that of the P―O group.In addition,it is very likely that Ca2+plays an important role in the interaction of BSAwith HA.It appears that the binding of Ca2+to the carbonyl-oxygen of the peptide bond in BSAcaused a significant,molecular,conformationalrearrangement of polypeptide backbones from β-pleated sheet to helical circles of α-helix and β-turn.This change appears to have been followed by much hydrogen of polypeptides being driven to bind PO43-and OH-effectively and much―C=O and H―N―groups of the peptide bond being freed from inter-chain hydrogenbonding to act on Ca2+and combine strongly with the HAsurface.This might reasonably be expected to promote hard tissue regeneration.BSA seems to be activated by the inductive effect of Ca2+via the molecular rearrangement of polypeptide backbones from pleated sheet to helical circles and in turn reacts strongly on the HA,resulting in profound effects on the course of biomineralization.
Biomaterial;Protein adsorption;Polypeptide;IR spectroscopy;FTIR-ATR;Biomineralization
July 2,2015;Revised:November 26,2015;Published on Web:November 30,2015.*Corresponding author.Email:stzylin@xmu.edu.cn;Tel:+86-592-2185956.
The project was supported by the National Natural Science Foundation of China(51571169).
国家自然科学基金(51571169)资助项目
©Editorial office ofActa Physico-Chimica Sinica
猜你喜欢
高中数理化(2022年14期)2022-08-15
唐山师范学院学报(2020年6期)2020-04-16
唐山师范学院学报(2019年3期)2019-06-18
原子与分子物理学报(2019年5期)2019-04-28
铜仁学院学报(2018年6期)2018-07-05
广州化学(2016年5期)2016-11-15
温州大学学报(自然科学版)(2016年1期)2016-10-27
中学化学(2015年12期)2016-01-19
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
应用化工(2014年7期)2014-08-09
