
自噬在肝稳态调节作用中的研究进展
2016-11-01 02:11黄慧李洪艳邹伟
生物工程学报 2016年9期
黄慧,李洪艳,邹伟
自噬在肝稳态调节作用中的研究进展
黄慧1,2,李洪艳1,2,邹伟1,2
1 辽宁师范大学生命科学学院,辽宁大连116081 2 辽宁省生物技术与分子药物研发重点实验室,辽宁大连 116081
肝脏是人体最大的消化腺,也是最主要的代谢器官。自20世纪60年代,人们在肝脏溶酶体的研究中提出“自噬”这一概念时,就发现肝脏内的营养水平与激素影响自噬活动。近年来的研究表明,自噬不仅是正常的生理过程,也参与许多病理过程的调节。本文介绍了自噬在健康肝脏中维持稳态的作用,旨在为肝脏生理学及自噬失调相关疾病的治疗提供新思路。
肝脏,稳态,自噬,代谢,调节
自噬 (Autophagy) 是细胞内的成分在溶酶体中降解的一系列分子反应[1]。错误合成的蛋白质或受到损伤的细胞器在产生毒性效应之前需要被清除[2]。自噬具有清除受损的细胞内成分或者对蛋白质进行质控的功能。事实上,即使胞内的蛋白质或细胞器没有受到损伤,它们还是会进行某种程度的合成和降解。这种过程能维持细胞的自我更新并防止细胞因受损而无法正常发挥作用。此外,通过自噬降解的大分子物质能够产生可以循环利用的氨基酸、游离的脂肪酸和部分碳水化合物,当细胞缺少营养时,它们可以重新合成新的细胞内成分或进一步氧化产生ATP[3]。因此,自噬是细胞生长、分化、存活和自我平衡的重要途径。
众所周知,肝脏是人体最大的消化腺,也是最重要的代谢器官。肝脏内的稳态依赖于大分子物质生物合成和分解代谢间的平衡。自噬就是细胞用来达到这种动态平衡的重要机制之一,它是普遍存在于大部分真核细胞中的一种现象,从酵母到人类存在着共同的自噬分子调控机制。有趣的是,它能够保护细胞,也能造成细胞损伤,在细胞的生长、发育和疾病中起着重要的作用。通过自噬来维持肝稳态对其功能的正常发挥至关重要。自噬既可以在生理条件下保护肝细胞,也可以在多种病理条件下影响肝细胞[4-6]。因此,从自噬的角度认识肝稳态,为了解肝病发生的机制提供新的思路。
1 肝细胞的自噬种类及特征
近年来大量文献显示自噬参与肝细胞稳态的维持和病变的发生过程[7-8]。人们将肝细胞自噬分为3种类型:巨自噬 (Macroautophagy),微自噬 (Microautophagy) 和分子伴侣介导的自噬 (Chaperone-mediated autophagy, CMA)。此外,根据激活方式的不同,自噬又可以分为“组成型”自噬 (持续激活) 和“诱导型”自噬 (受到刺激后激活)。代谢物质被大量的随机分配或选择性的 (被运送分子具有特异性靶点)运送至溶酶体中降解。
巨自噬是自噬中最主要的也是研究的最为广泛的类型。巨自噬中,吞噬小泡能将胞质内的可溶性物质和细胞器包绕形成自噬小体,一旦自噬小体与溶酶体融合形成自噬溶酶体,就会将胞质内成分隔离并降解,促进氨基酸和脂肪酸等小分子物质的循环利用 (图1)。自噬小体的形成也是巨自噬区别于其他途径的特别之处。巨自噬能参与调节正常细胞的代谢过程,而且在肝癌细胞的营养供应中发挥至关重要的作用[9]。巨自噬能够降解特定的细胞器,如线粒体,过氧化物酶体和脂质体等。在巨自噬过程的不同阶段,大约有30多种自噬相关基因表达的蛋白产物组装成功能性蛋白复合物协同发挥作用[10-11]。巨自噬的最后一步与其他途径相同,即将溶酶体降解循环的产物运送至细胞质中。Yang等早在2007年就在酵母菌中鉴定了位于溶酶体膜上参与循环过程的通透酶,但是这些酶的同系物在哺乳动物中的鉴定研究才刚刚开始[12]。Zoncu等2011年发现,通过调节参与自噬活动的复合物能够检测到溶酶体中释放的氨基酸和糖类等物质。例如,哺乳动物细胞中,起负调控作用的雷帕霉素 (mTOR) 靶点就是巨自噬中最有特征的因子,它位于溶酶体的表面,受自噬内涵体释放的氨基酸和葡萄糖调控[13]。
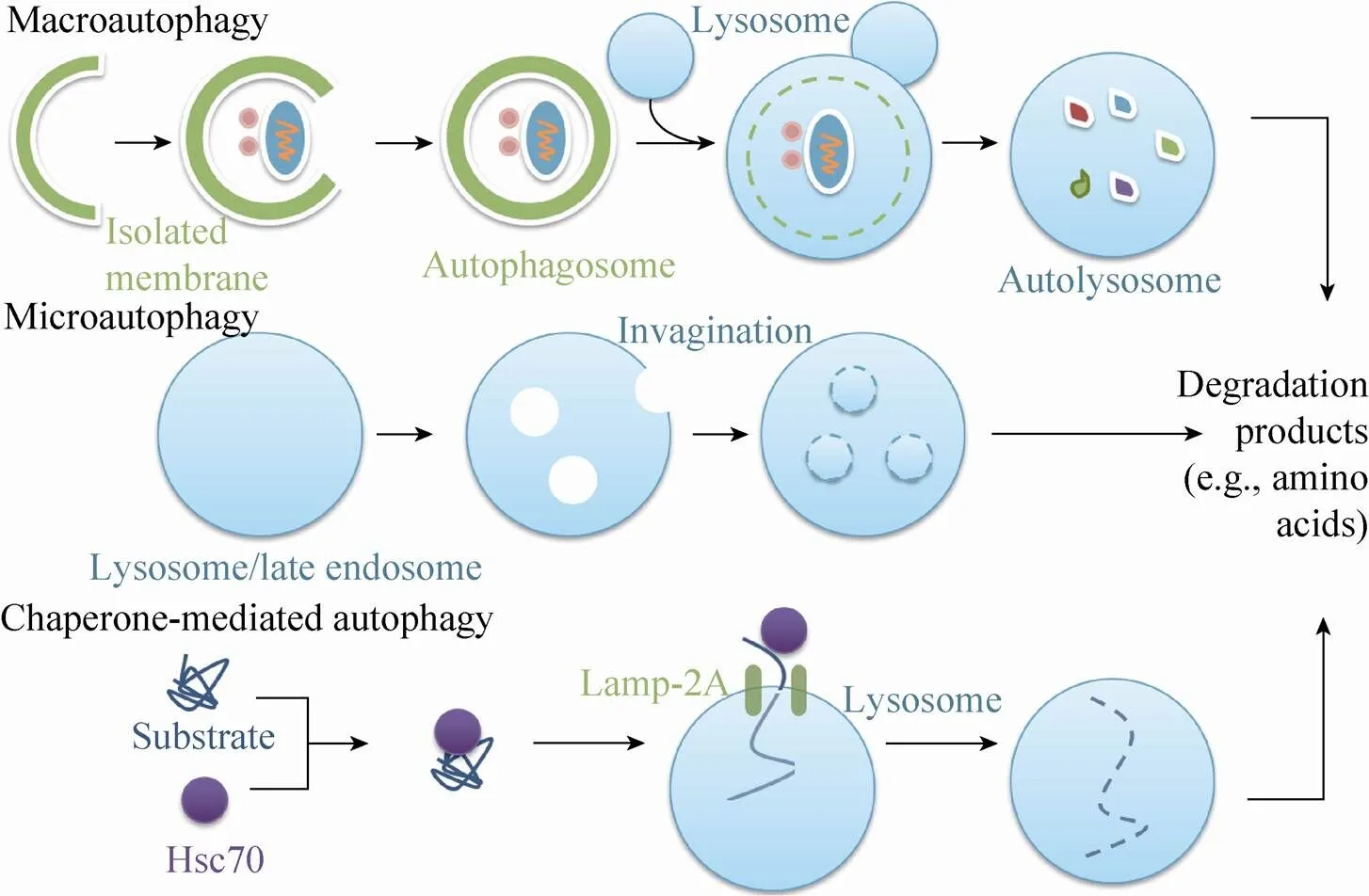
图1 肝细胞的自噬类型
最初,人们认为巨自噬只有在饥饿等应激条件下才会被激活。然而,在非应激条件下,巨自噬同样能对肝脏产生影响。由于巨自噬过程在基因水平被阻断,从而导致受损细胞器和蛋白质的积累,因此肝细胞稳态的维持需要巨自噬过程持续发生[14]。在大多数器官中,不同的刺激 (如内质网应激,缺氧,氧化应激,DNA损伤等) 可以通过不同的信号通路激活巨自噬途径,其中包括JNK、CaMKK、LKB、AKt、Sirt1、PERK、PDGF、AMPK以及p53介导的信号通路[15]。
微自噬发生时,溶酶体膜内陷将待降解的物质包裹并内化[16](图1)。早在1998年,Sakai等研究发现,参与巨自噬过程的基因都是高度保守的,与之不同的是,酵母菌中参与微自噬的基因在哺乳动物中并不保守[17],那么哺乳动物中是否发生了微自噬过程,这种保守性的缺失引起了人们的质疑。然而,2011年,Sahu等研究发现,类似于微自噬的过程在肝细胞等哺乳动物细胞的晚期内涵体中发生[18]。内涵体发生微自噬需要ESCRT (endosomal sorting complex required for transport, ESCRT) 复合物参与形成内吞小泡,以完成质膜蛋白的内化和循环[18]。尽管微自噬过程在肝脏中首先被发现,但是肝细胞微自噬的分子机制目前仍不清楚。
肝自噬的第3种类型是分子伴侣介导的自噬 (CMA),与其他方式相比,CMA具有不同的特点,即分子伴侣[19]在胞浆中识别底物蛋白并通过复杂的易位跨膜将底物运送至溶酶体中,自噬过程中不涉及膜重组过程。Cuervo等研究发现[20-21],CMA能将哺乳动物细胞中30%含有KFERQ基序的蛋白运送到溶酶体中,在转运过程中,底物蛋白需要进行折叠,折叠后的蛋白才能穿过溶酶体双层膜。其中LAMP-2A和持续表达的分子伴侣Hsc70在CMA中发挥重要作用[20,22-23]。分子伴侣Hsc70能特异性的识别胞质内含有KFERQ基序的蛋白,折叠后的蛋白通过多聚复合物被运送至溶酶体,与溶酶体膜上的受体蛋白Lamp-2A结合,随后被降解 (图1)。CMA最初在体外培养的成纤维细胞中被鉴定,但是CMA对肝稳态的调节却是十分重要的。当肝细胞处于长期营养剥夺、氧化应激和蛋白毒性等不同损伤时,CMA作为细胞反应的一部分表达上调[24-25]。Codogno等新近发现,CMA能调控由营养状态改变或衰老引起的受损蛋白质的降解,揭示CMA在脂类和碳水化合物代谢过程中发挥重要的作用[26]。但是,到目前为止,其分子机制尚未可知。
2 自噬在肝稳态调节中的作用
自从de Duve发现饥饿和胰高血糖素能引起肝细胞自噬这一现象开始,细胞代谢就和自噬过程密切相关[27]。对有机体来说,肝脏对代谢稳态的维持和能量平衡发挥着至关重要的作用。新近研究表明,肝细胞自噬过程被破坏会引起肥胖、脂肪肝等代谢失调相关疾病[28]。因此,了解肝细胞自噬调控肝脏代谢的分子机制显得尤为重要。近年研究发现,自噬主要通过如下3种机制帮助肝脏维持稳态。
2.1 自噬在维持肝细胞能量平衡中的作用
Ezaki等研究发现,在处于饥饿状态的前4−6 h,肝细胞中的巨自噬就被广泛激活。溶酶体降解蛋白质产生的氨基酸可进入三羧酸循环用于ATP的产生或糖异生过程[7]。对于新生儿来说,在由胎盘输送营养和泌乳期之间的这段关键时期,自噬过程的诱导对新生儿适应饥饿环境非常重要。而对于肝脏中自噬基因缺陷型小鼠来说,由于饥饿诱导的蛋白质水解和脂类水解不能发生,所以它们不具备适应饥饿的能力,从而导致新生小鼠能量代谢失衡[14]。这种失衡进一步受到线粒体功能的威胁,线粒体对脂质的β氧化作用至关重要,然而由于自噬引起细胞器转换效率降低,使线粒体的功能受到损伤[14]。
循环中糖原的增多以及胰岛素和氨基酸水平的降低能够在饥饿状态下促进肝脏巨自噬过程的激活,而且糖原、胰岛素和氨基酸是肝脏巨自噬过程有效的抑制剂[27]。当饥饿8 h时,肝脏中巨自噬降解的蛋白质减少,同时逐渐激活CMA过程,该过程能在营养剥夺24 h时达到最佳状态并持续激活3 d,此时它作为氨基酸的内源性来源发挥作用[29]。2014年,Schneider等在缺失CMA模型的研究中发现,在饥饿期间若不能激活CMA途径,会导致肝脏代谢的能量失衡[21]。对于饥饿诱导CMA的机制目前尚未可知,但是,视黄酸受体α (RABα) 介导的信号通路是目前调节CMA活动的唯一途径[30]。
尽管到目前为止,人们对蛋白质降解的研究最为深入,但是近年来,自噬对肝脏内以其他形式储存能量的物质的降解也引起了人们的广泛关注。溶酶体上含有不同类型的水解酶,不仅能降解蛋白质,还能降解碳水化合物,脂质和核酸。当机体不能通过正常饮食摄取葡萄糖时,肝脏中储存的糖原就成为了葡萄糖的重要来源。Hers早在1963年发现,在饥饿期间,糖原通过巨自噬或微自噬的方式被运送至溶酶体,被溶酶体上的酸性α-葡萄糖苷酶降解[31],这个过程也被称为糖自噬 (Glycophagy)。Stbd1蛋白的发现强化了人们对糖自噬的认识。Stbd1是底物识别受体,它通过与自噬相关蛋白GABARAPL1相互作用从而将糖原运送至自噬小体[32]。然而,细胞感知葡萄糖水平下降并选择糖自噬的机制目前仍不清楚。早在2009年,Takikita等发现,在酵母菌的液泡中,巨自噬也参与了糖原的运送和降解过程[33],但是哺乳动物中是否也存在这一过程目前尚未可知。新近研究表明,cAMP和mTOR信号通路都能够调节新生肝脏糖自噬的过程[34],并且生长激素和肝脏葡萄糖调节之间存在一定联系[35]。
近年来,溶酶体对肝脏脂质的降解获得了同样的关注。Singh等曾经观察到,饥饿8 h肝细胞中持续发生了巨自噬过程,但是从自噬小体中分离出的物质是脂质[36]。脂质存在于自噬小体内并被运送至溶酶体,通过选择性的方式被溶酶体内的水解酶分解,此后称这种方式为巨噬脂 (Lipophagy)[36]。溶酶体介导的脂解是脂肪分解过程的一个分支,它是由胞质腔内的酯酶来完成的,不依赖于溶酶体内的脂肪酶。O'Rourke等指出,饥饿激活了肝细胞内一个完整的转录过程,以为自噬/溶酶体系统完成脂类货物的运送和加工做好准备[37]。尽管溶酶体中的脂类自噬在全部脂解的正常水平以下,但是它能够阻止肝脏中的脂肪大量积累,从而防止由日常膳食引起的脂肪负荷[36]。牛黄熊去氧胆酸 (Tauroursodeoxycholic acid, TUDCA) 是肥胖时能量代谢的重要调节因子,Guo等新近发现,注射TUDCA的肥胖小鼠体重减少,血糖降低,对胰岛素的敏感性增加。此外,TUDCA还能使肥胖小鼠肝脏中异常的自噬功能恢复正常,改善肥胖小鼠的糖脂代谢失衡[38]。有趣的是,由巨自噬介导的脂肪转移不只在肝细胞中发生。脂类自噬对肝脏星形细胞的激活也至关重要,是肝脏纤维化的关键过程之一[8],因此可以通过瞬时阻断自噬过程的方法为肝脏纤维化的治疗提供新的策略。
2.2 自噬参与线粒体更新
线粒体是细胞内ATP的主要来源,肝细胞能量平衡的维持与线粒体功能密切相关。当受到活性氧损伤时,细胞内的多种结构会发生改变,其中线粒体最为严重,通过改变自噬的方式可以使其降解,这一过程又称为线粒体自噬 (Mitophagy)[39]。线粒体自噬可以防止ATP合成酶生成ATP,同时可以限制自由基的产生。通过这种方式恢复能量平衡,可以阻止细胞死亡途径被激活从而减少细胞损伤。
研究表明,线粒体自噬过程中拥有识别和清除共同存在的机制[40-41]。例如,PINK1/PARKIN信号通路在线粒体膜去极化之后被激活,当膜受到损伤后会导致PINK1积聚在线粒体膜表面,将泛素连接酶PARKIN招募到膜上。被PARKIN泛素化的蛋白作为ATG识别和线粒体自噬的标记[42],PARKIN引起的蛋白质降解能防止线粒体成球状积累,在损伤的肝脏中能观察到受损的线粒体分离的现象[43]。当自噬过程受到破坏时,溶酶体能直接与球状线粒体融合,以作为线粒体降解后的替代机制[44]。Wu等新近指出,在缺氧和营养状态发生改变时,其他的受体,如BCL2、BNIP3、NIX和FUNDC1等同样会激活线粒体自噬过程[44]。线粒体自噬存在不同的类型,哪些特定的类型更倾向于参与细胞质控,哪些类型更易于改变线粒体功能,未来关于线粒体自噬不同类型之间的比较研究能帮助人们更深入地理解这些问题。
2.3 自噬参与肝细胞酶的降解
自噬除了能够分解储存的能量之外,还可以通过降解肝细胞代谢过程中的酶来调节肝细胞的能量平衡。然而,由于CMA能够靶向的降解单一的蛋白质,因此在调节肝细胞酶的降解过程中CMA具有重要作用。事实上,糖酵解的关键酶已经通过选择性自噬途径进行了蛋白质水解[29-45]。在细胞代谢过程中,通过巨自噬降解的调节蛋白在肝脏代谢的转录调控中也起到了一定作用,比如p62[46]。在肝脏缺陷型小鼠中,由于p62蛋白的积累,导致部分的代谢过程受到了破坏,因为与野生型小鼠相比,缺失p62的小鼠呈现出丧失自噬过程的表型[47]。
自噬和酶水解之间的关系是在肿瘤细胞代谢过程中被发现的。近年来的研究证明了CMA和肿瘤代谢之间复杂的相互作用。本室前期研究发现,胞膜窖标志蛋白窖蛋白-1 (Caveolin-1,Cav-1) 与肿瘤细胞的自噬密切相关[48]。Cav-1的缺乏促进自噬标记物的表达。例如,让hTERT-fibroblasts (人类无限增殖的成纤维细胞) 经受缺氧,用Cav-1的抗体或自噬标记物(LC3A/B和ATG16L) 的抗体和线粒体自噬的标记物(BNIP3和BNIP3L) 抗体标记,结果表明,在纤维母细胞中,低氧诱导的自噬和线粒体自噬标记物的表达,与Cav-1的下调有直接关 联[49]。另外,Cav-1在自噬标志蛋白LC3B引起的肺气肿细胞凋亡过程中,起着非常重要的作用[50]。同时,本实验室新近研究发现,通过全基因组表达谱芯片的方法研究肝癌细胞自噬前后的基因表达变化,结果显示,自噬后增殖凋亡相关的信号通路PI3K/Akt发生差异表达变化,说明自噬可能通过影响PI3K/Akt及相关信号通路参与肝癌细胞的代谢调控 (结果尚未发表)。Kon等研究发现,在超过14种类型的肿瘤中,CMA均上调,阻断CMA途径后肿瘤生长速度减慢,并且显著降低了肿瘤的转移频率[51]。
尽管在肿瘤细胞中首先发现CMA和细胞代谢之间的关系,但是Schneider等新近研究表明,哺乳动物肝细胞中,在正常状态和饥饿条件下阻断CMA途径对参与碳水化合物和氨基酸代谢过程中酶的调节具有重要的生理作 用[13]。Schneider等从肝脏中分离出多种的溶酶体,它们参与不同类型的CMA过程,对这些溶酶体进行蛋白组学的比较分析,结果表明,超过40%的蛋白质作为CMA的底物参与代谢过程,其中约2/3的蛋白质参与碳水化合物和脂类代谢[21]。由于肝脏无法进行CMA过程,因此糖酵解相关酶的表达上调,如GAPDH、PK、醛缩酶A、苹果酸脱氢酶1或烯醇化酶1,因此会导致肝脏的基础糖酵解水平升高。肝脏持续消耗葡萄糖会导致糖异生过程的缺失和肝糖原的缺乏。而且在饥饿条件下,CMA的缺失尤为明显,此时,CMA会降解糖酵解相关酶,以减少肝脏中葡萄糖的消耗。
当肝脏不能进行CMA时,糖酵解持续发生会造成机体的能量失衡,同时会引起周围脂肪组织的损耗[21]。如果未能及时调动肝脏中的脂类会引起部分周围脂肪组织的减少。脂肪生成酶被CMA降解后表达上调,尽管这能够显著促进缺失CMA肝脏中的脂肪变性,但是,Kaushik等新近研究表明,脂肪积累主要是由于脂肪不能被及时分解所导致的[52]。在这种情况下,为了控制脂肪的分解过程,CMA的作用底物由酶转变成肝脏中覆盖在脂滴表面的蛋白,即周脂素2 (Perilipin 2) 和周脂素3 (Perilipin 3)。在脂解过程中,需要移除一部分周脂素以允许胞质中的自噬相关蛋白和脂肪酶进入脂滴中心完成巨噬脂和脂解过程。移除周脂素的过程需要Hsc70的识别和磷酸化过程,随后将它们运送到溶酶体中降解[52]。如果CMA过程中溶酶体上的受体是消融的,Hsc70和周脂素仍能坚持与脂滴表面结合,以阻止脂解过程的发生。CMA对周脂素的降解可以在饥饿状态下发生,以动员肝脏脂质被周围组织利用,也可以在脂肪持续生成时发生,以防止过度摄食对肝脏产生毒性效应[52]。
综上所述,CMA过程对肝脏稳态的维持至关重要,同时它是肝脏适应营养变化时的重要组成部分。
3 小结与展望
自噬不仅能在饥饿时为机体提供能量,它与肝脏生理之间的复杂联系更为重要。自噬还具有一系列新功能,其中包括对肝脏代谢的整体调控以及对蛋白质和细胞器的质控。肝细胞自噬的复杂性不仅因为它在不同的生理过程中发挥不同的作用,还因为有多重机制参与该过程的调节。因此,需要进一步研究自噬在不同生理过程中的信号转导机制。靶向改变相关疾病中的自噬途径,将为临床上肝脏自噬失调相关疾病的治疗提供新思路。
REFERENCES
[1] Zhang H, Baehrecke EH. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol, 2015, 25(7): 376–387.
[2] Madrigal-Matute J, Cuervo AM. Regulation of liver metabolism by autophagy. Gastroenterology, 2016, 150(2): 328–339.
[3] Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab, 2011, 13(5): 495–504.
[4] Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun, 2015, 6: 6823.
[5] Jiang XJ, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest, 2015, 125(1): 47–54.
[6] Schneider JL, Villarroya J, Diaz-Carretero A, et al. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell, 2015, 14(2): 249–264.
[7] Ezaki J, Matsumoto N, Takeda-Ezaki M, et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy, 2011, 7(7): 727–736.
[8] Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology, 2012, 142(4): 938–946.
[9] Feng YC, He D, Yao ZY, et al. The machinery of macroautophagy. Cell Res, 2014, 24(1): 24–41.
[10] Yang ZF, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol, 2010, 22(2): 124–131.
[11] Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol, 2011, 27: 107–132.
[12] Yang ZF, Klionsky DJ. Permeases recycle amino acids resulting from autophagy. Autophagy, 2007, 3(2): 149–150.
[13] Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science, 2011, 334(6056): 678–683.
[14] Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in-deficient mice. J Cell Biol, 2005, 169(3): 425–434.
[15] Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell, 2010, 40(2): 280–293.
[16] Mortimore GE, Hutson NJ, Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci USA, 1983, 80(8): 2179–2183.
[17] Sakai Y, Koller A, Rangell LK, et al. Peroxisome degradation by microautophagy in: identification of specific steps and morphological intermediates. J Cell Biol, 1998, 141(3): 625–636.
[18] Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell, 2011, 20(1): 131–139.
[19] Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci, 1990, 15(8): 305–309.
[20] Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol, 2012, 22(8): 407–417.
[21] Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab, 2014, 20(3): 417–432.
[22] Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacol Res, 2012, 66(6): 484–493.
[23] Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol, 2011, 23(2): 184–189.
[24] Kiffin R, Christian CJ, Knecht E, et al. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell, 2004, 15(11): 4829–4840.
[25] Massey AC, Kaushik S, Sovak G, et al. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Nat Acad Sci USA, 2006, 103(15): 5805–5810.
[26] Codogno P, Lotersztajn S. When autophagy chaperones liver metabolism. Cell Metab, 2014, 20(3): 392–393.
[27] Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol, 1967, 35(2): C11–C16.
[28] Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol, 2014, 11(3): 187–200.
[29] Cuervo AM, Knecht E, Terlecky SR, et al. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol, 1995, 269(5 Pt 1): C1200–C1208.
[30] Anguiano J, Garner TP, Mahalingam M, et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol, 2013, 9(6): 374–382.
[31] Hers HG. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe's disease). Biochem J, 1963, 86: 11–16.
[32] Jiang SX, Heller B, Tagliabracci VS, et al. Starch binding domain-containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J Biol Chem, 2010, 285(45): 34960–34971.
[33] Takikita S, Myerowitz R, Schreiner C, et al. The values and limits of anmodel of Pompe disease: the best laid schemes o' mice an' men. Autophagy, 2009, 5(5): 729–731.
[34] Kondomerkos DJ, Kalamidas SA, Kotoulas OB, et al. Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol Histopathol, 2005, 20(3): 689–696.
[35] Zhang YY, Fang F, Goldstein JL, et al. Reduced autophagy in livers of fasted, fat-depleted, ghrelin-deficient mice: reversal by growth hormone. Proc Natl Acad Sci USA, 2015, 112(4): 1226–1231.
[36] Singh R, Kaushik S, Wang YJ, et al. Autophagy regulates lipid metabolism. Nature, 2009, 458(7242): 1131–1135.
[37] O'Rourke E J, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol, 2013, 15(6): 668–676.
[38] Guo QY, Shi QD, Li HX, et al. Glycolipid metabolism disorder in the liver of obese mice is improved by TUDCAthe restoration of defective hepatic autophagy. Int J Endocrinol, 2015, 2015: 687938.
[39] Kim I, Rodriguez-Enriquez S, Lemasters J J. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys, 2007, 462(2): 245–253.
[40] Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol, 2014, 2: 749–754.
[41] Zhu JH, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy, 2013, 9(11): 1663–1676.
[42] Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron, 2015, 85(2): 257–273.
[43] Ding WX, Guo FL, Ni HM, et al. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem, 2012, 287(50): 42379–42388.
[44] Wu H, Chen Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid Redox Signal, 2015, 22(12): 1032–1046.
[45] Lv L, Li D, Zhao D, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell, 2011, 42(6): 719–730.
[46] Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell, 2009, 137(6): 1062–1075.
[47] Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell, 2007, 131(6): 1149–1163.
[48] Liu Y, Wang Y, Shi D, et al. Autophagy and caveolin-1 in cancer: a review. Chin J Biotech, 2012, 28(8): 912−917 (in Chinese).刘艳, 王洋, 史丹, 等. 肿瘤细胞的自噬和窖蛋白-1. 生物工程学报, 2012, 28(8): 912−917.
[49] Erez N, Truitt M, Olson P, et al. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell, 2010, 17(2): 135−147.
[50] Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA, 2010, 107(44): 18880−18885.
[51] Kon M, Kiffin R, Koga H, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med, 2011, 3(109): 109–117.
[52] Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol, 2015, 17(6): 759–770.
(本文责编 陈宏宇)
Progress of autophagy in regulating liver homeostasis
Hui Huang1,2, Hongyan Li1,2, and Wei Zou1,2
1,,116081,,2,116081,,
Liver is the largest human digestive gland and the most important metabolic organ. When autophagy was proposed during studying liver lysosomes in the 1960s, it was found that nutrient levels and hormones could influence autophagy activity. Recent studies show that autophagy is not only normal physiological processes, but also involved in the regulation of many pathological processes. This article summarizes the role of liver autophagy in the maintenance of homeostasis in the healthy liver, and provides new ideas for liver physiology and treating diseases associated with autophagy disorders.
liver, homeostasis, autophagy, metabolism, regulation
December 30, 2015; Accepted: February 26, 2016
Wei Zou. Tel/Fax: +86-411-8582-7080; E-mail: weizou60@126.com
Supported by:National Natural Science Foundation of China (No. 30570225), Liaoning Science and Technology Project (No. 2015020568).
国家自然科学基金 (No. 30570225),辽宁省科学技术计划项目 (No. 2015020568) 资助。
网络出版时间:2016-03-11 网络出版地址:http://www.cnki.net/kcms/detail/11.1998.Q.20160311.1119.001.html
黄慧, 李洪艳, 邹伟. 自噬在肝稳态调节作用中的研究进展. 生物工程学报, 2016, 32(9): 1185–1193.
Huang H, Li HY, Zou W. Progress of autophagy in regulating liver homeostasis. Chin J Biotech, 2016, 32(9): 1185–1193.
猜你喜欢
大电机技术(2022年3期)2022-08-06
核科学与工程(2021年4期)2022-01-12
金桥(2021年10期)2021-11-05
煤气与热力(2021年4期)2021-06-09
生物化工(2021年2期)2021-01-19
生物化工(2020年1期)2020-02-17
中华戏曲(2020年1期)2020-02-12
读与写(2019年35期)2019-11-05
小学生作文(低年级适用)(2019年3期)2019-04-04
现代职业教育·高职高专(2018年7期)2018-05-14
