
20-HETE对大鼠心肌缺血再灌注损伤中NADPH氧化酶活性及ROS生成的影响*
2016-11-01 03:02郭立荣刘万力赵红艳
遵义医科大学学报 2016年4期
韩 勇,郭立荣,贺 滟,刘万力,王 梅,赵红艳,何 娅
(1.遵义医学院 基础医学院生理学教研室,贵州 遵义 563099;2. 遵义医学院 基础医学院形态学实验室,贵州 遵义 563099)
基础医学研究
20-HETE对大鼠心肌缺血再灌注损伤中NADPH氧化酶活性及ROS生成的影响*
韩勇1,郭立荣2,贺滟1,刘万力1,王梅1,赵红艳1,何娅1
(1.遵义医学院 基础医学院生理学教研室,贵州 遵义563099;2. 遵义医学院 基础医学院形态学实验室,贵州 遵义563099)
目的 探讨20-HETE加重大鼠离体心肌缺血再灌注损伤的作用及机制。方法 雄性Wistar大鼠随机分为正常对照组(Con组),缺血再灌注模型组(IR组),IR+20-HETE合成酶抑制剂(HET0016)组,Con+HET0016组,每组12只,通过Langendorff灌流系统建立大鼠离体心肌缺血再灌注模型,全心缺血35 min再灌注40 min,PowerLab/8P系统实时监测心肌力学指标;灌流结束后取心肌组织TTC法检测心肌梗死面积;DHE荧光探针法检测活性氧簇(reactive oxygen species,ROS)含量;Western blot检测NADPH氧化酶亚基p47phox磷酸化水平;细胞色素C的还原法检测NADPH氧化酶活性。结果 离体心脏缺血再灌注后,加入20-HETE合成酶抑制剂HET0016(1 μmol/L)后明显改善了IR导致的心肌力学指标下降,LVDP由IR组的(48.6±3.4)%升至(71.7±3.5)%,+dP/dtmax由(51.9±2.1)%升至(69±3.2)%,-dP/dtmax由(47.1±3.6)%升至(64.1±3.8)%(P<0.05);IR+HET0016组心肌梗死面积比IR组减小了37.2%(P<0.05)。DHE荧光探针检测发现IR+HET0016处理组心肌组织中ROS含量比IR组降低了45.8%(P<0.05);最后通过对NADPH氧化酶活性检测发现,HET0016显著减少了IR引起的NADPH氧化酶p47phox亚基磷酸化水平及全酶活性的升高(P<0.05)。结论 20-HETE通过激活NADPH氧化酶诱导过量ROS的生成,加重心肌缺血再灌注损伤。
20-羟二十烷花生四烯酸;大鼠;心肌缺血再灌注损伤;活性氧簇;NADPH氧化酶
20-羟二十烷花生四烯酸(20-hydroxyeicosatetraenoic acid, 20-HETE)是由细胞色素P-450(ω-羟化酶)催化花生四烯酸(arachidonic acid, AA)生成的内源性活性物质,目前被认为是调节肾、脑等小动脉血管紧张度的重要因子[1-3],但其对心脏功能的影响研究甚少。近年来有研究报道,在大鼠和犬的心脏中鉴定出CYP4A和4F表达,在心肌缺血再灌注损伤中,20-HETE水平明显增加,心肌组织中CYP 4A、4F表达和活性显著增强,应用CYP ω-羟化酶抑制剂,如17 ODYA和DDMS,可改善再灌注后的心肌功能恢复,减少心肌梗死面积[4-5]。上述研究表明,20-HETE参与到了心肌缺血再灌注当中,加重了再灌注后心肌损伤,但是其中确切的分子机制仍不明确。
在心肌缺血再灌注中,活性氧簇(reactive oxygen species,ROS)发挥了至关重要的作用,被认为是引起心肌缺血再灌注损伤的主要启动因子[6-7],但ROS生成的来源及具体机制尚待深入研究。因此,本研究的目的是在大鼠离体心脏缺血再灌注模型上,研究20-HETE对心肌功能及ROS生成的影响和机制,从而探究20-HETE加重再灌注后心肌损伤的机制。
1 材料与方法
1.1实验动物及试剂雄性Wistar大鼠(220±20)g随机分为正常对照组(Con组),缺血再灌注模型组(IR组),IR+20-HETE合成酶抑制剂(HET0016)组,Con+HET0016组,每组12只,实验前标准饮食饲养1周。
20-HETE购于Sigma公司,ROS检测试剂盒及NADPH氧化酶活性检测试剂盒购于南京建成生物制品公司,特异性p47phox和磷酸化p47phox抗体、HRP标记的二抗购自Santa生物技术公司,其他试剂均采用化学分析纯。
1.2方法
1.2.1动物模型建立大鼠使用乌拉坦(1 g/kg)腹腔注射麻醉,开胸取出心脏置于4 ℃的KH液(含肝素,50 U/mL)中修剪,将主动脉套管后置于Langendorff心脏灌流系统上,37 ℃下使用KH液(NaCl 118.0 mmol/L, NaHCO325.0 mmol/L, KCl 4.7 mmol/L, KH2PO41.2 mmol/L, MgSO4·7H2O 1.2 mmol/L, CaCl22.0 mmol/L, glucose 11.0 mmol/L)进行主动脉逆行灌流。经大鼠左心房插入左心室一个充水小球囊,另一端连接到压力传感器监测心室内压变化。使用Powerlab/8P系统连续记录左心室发展压(LVDP),左心室舒张末期压(LVEDP),左心室内压最大上升/下降速率(±dP/dtmax)。
1.2.2心肌缺血再灌注实验方案37 ℃条件下,离体心脏缺血前灌流20 min平衡,HET0016组心脏在缺血前10 min灌流HET0016(1 μmol/L),然后全心缺血35 min,再灌注15 min后检测ROS水平,或者再灌注40 min后用于检测NADPH氧化酶活性,或者再灌注120 min用以检测心肌梗死面积。PowerLab/8P系统实时检测缺血前及再灌后的左心室功能及血流动力学变化。
1.2.3心梗面积测定再灌注后的心脏在-20 ℃下冷冻1 h,切片(2~3 mm)放入含有1% TTC的磷酸盐缓冲液中,37 ℃条件下孵育15 min染色,10%甲醛固定。梗死区通过测定染色区域(红色,存活组织)和非染色区域(白色,坏死组织)面积占全部左心室面积比值后进行比较。
1.2.4心肌ROS水平测定ROS的生成使用对氧敏感的荧光探针DHE法测定,离体心脏再灌注15 min后,剪成100 mm3的小块置入组织包埋剂中,液氮冷冻,冰冻切片(10 μm)后置于载玻片上,然后将切片在PBS缓冲液中使用DHE (1.6 μmol/L) 37 ℃避光孵育30 min,激光共聚焦显微镜下在488 nm激发光,560~660 nm滤波接收,获取荧光照片。
1.2.5NADPH氧化酶p47phox亚基磷酸化水平测定离体心脏再灌注后,取大鼠左心室样品(非梗死区)约100 mg,液氮内研磨,加入全细胞裂解液及蛋白酶抑制剂,用匀浆机充分匀浆,冰上处理30 min,4 ℃ 12 000 g 离心15 min,取上清BAC法蛋白质定量。NADPH氧化酶p47phox亚基磷酸化水平采用Western blot法测定,组织提取蛋白使用梯度SDS聚丙烯酰胺凝胶电泳分离,半干转仪将凝胶中蛋白转移至PVDF膜上,5%脱脂奶粉封闭2 h,分别加入一抗p47phox(1∶1 000)、磷酸化p47phox(1∶1 000),4 ℃孵育过夜。PBS洗膜后加入HRP标记二抗(羊抗鼠 IgG-HRP,1∶2 000),37 ℃孵育1 h,ECL检测试剂盒曝光,Quantity One蛋白条带进行分析。
1.2.6NADPH氧化酶活性检测NADPH氧化酶活性检测采用对超氧化物歧化酶(SOD)敏感的NADPH氧化酶对细胞色素C的还原法,由心肌组织提取的全蛋白(终浓度1 mg/mL,100 μL)中加入细胞色素C(500 μmol/L)和NADPH(100 μmol/L),分别在加入和不加入SOD(200 U/mL)情况下,室温孵育30 min,550 nm波长下检测吸光度,过氧化物产物含量(nmol/mg protein)通过不同吸光值进行比较计算,高铁细胞色素C到亚铁细胞色素C变化的摩尔吸光系数(ε) 21 000 M-1cm-1。

计,P<0.05被认为具有统计学意义。
2 结果
2.1HET0016对离体心脏缺血再灌注后心肌力学的影响应用Powerlab/8P系统连续实时记录大鼠离体心脏灌流过程中各组心脏血流动力学功能改变。与正常对照Con组相比,再灌注末期IR组心脏的各项心肌力学指标出现显著下降,左心室发展压(LVDP)下降至(48.6±3.4)%,左心室舒张末期压(LVEDP)升高至(41.8±3.3)%,左心室内压最大上升/下降速率(±dP/dtmax)分别降低至(51.9±2.1)%和(47.1±3.6)%(P<0.05)。缺血前10 min灌注20-HETE合成酶抑制剂HET0016(1 μmol/L)后,相比于IR组再灌注末期心肌力学指标明显改善,LVDP上升至(71.7±3.5)%,LVEDP降至(32.1±4.5)%,±dP/dtmax分别提高至(69.0±3.2)%和(64.1±3.8)%(P<0.05)。Con对照组中加入HET0016后对心肌力学指标影响不明显(P>0.05,见图1,表1)。
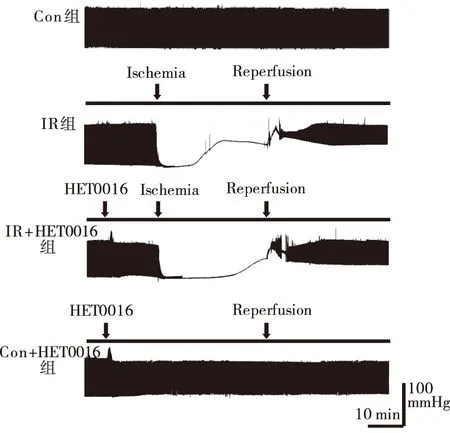
图1 心脏灌流实时心肌功能记录


组别缺血再灌注后(占缺血前平衡期百分比%)LVDPLVEDP+dP/dtmax-dP/dtmaxCon组95.0±2.35.4±1.493.3±1.989.4±1.8IR组48.6±3.4*41.8±3.3*51.9±2.1*47.1±3.6*IR+HET0016组71.7±3.5#32.1±4.5#69.0±3.2#64.1±3.8#Con+HET0016组93.9±3.36.2±2.491.1±3.990.3±2.7
*:P<0.05,与Con组相比;#:P<0.05,与IR组相比。
2.2HET0016对离体心脏缺血再灌注后心肌梗死面积的影响离体心脏全心缺血35 min再灌注120 min后,通过TTC染色法检测心肌梗死面积。与正常对照组相比,再灌注后IR组心肌梗死面积达到了(39.8±2.3)%,而加入HET0016组有效减少了由于IR引起的梗死面积增加,心肌梗死减少至(29.6±2.4)%(P<0.05)。Con+HET0016组心肌梗死面积与正常对照组相比变化不明显(P>0.05,见图2)。
2.320-HETE对再灌注后心肌组织ROS生成的影响研究认为ROS是引起缺血再灌注损伤的主要启动因子,但其生成的来源及具体机制尚待深入研究。因此本研究应用DHE荧光探针法检测了HET0016对心肌再灌注后ROS生成的影响。与正常对照组相比,再灌注后IR组荧光强度由19增加至105;缺血前灌流液中加入HET0016(1 μmol/L)后心肌组织荧光强度为72,显著低于IR组(P<0.05);Con+HET0016组心肌组织荧光强度较正常对照组无明显变化(P>0.05,见图3)。
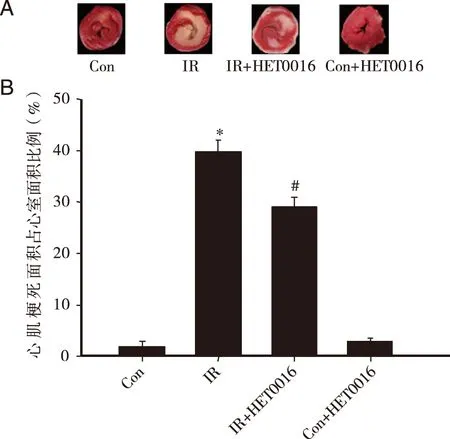
A:TTC染色法检测所得心肌梗死面积;B:心肌梗死面积统计分析;*:P<0.05,与Con组相比;#:P<0.05,与IR组相比。 图2 HET0016对离体心脏缺血再灌注后心肌梗死面积的影响

A:各组心肌组织荧光照片(200×);B:荧光值统计分析;*:P<0.05,与Con组相比;#:P<0.05,与IR组相比。图3 HET0016对再灌注后心肌组织ROS生成的影响
2.4HET0016对再灌注后NADPH氧化酶亚基p47phox磷酸化及全酶活性的影响有研究表明心肌缺血再灌注损伤中ROS过量生成与NADPH氧化酶激活有关,NADPH氧化酶由多种亚基构成,其中p47phox亚基磷酸化是其被激活的关键步骤,因此本研究应用Western Blot方法检测了HET0016对NADPH氧化酶亚基p47phox磷酸化的作用,采用细胞色素C还原法检测NADPH氧化酶活性。结果发现,与正常对照组相比,缺血再灌注后的IR组心肌组织中磷酸化的p47phox亚基由(22.5±2.3)%增加至(53.5±4.4)%;而当应用HET0016阻断该进程中20-HETE合成后,心肌组织中磷酸化的p47phox亚基减少至(32.7±5.3)%,显著低于IR组(P<0.05);通过对NADPH氧化酶全酶活性检测发现类似的结果,再灌注后IR组心脏NADPH氧化酶活性由正常对照组的11.2升高至18.3;HET0016组心脏NADPH氧化酶活性降低至13.9,显著低于IR组(P<0.05);Con+HET0016组心肌组织中p47phox亚基磷酸化水平及NADPH氧化酶活性无明显变化(P>0.05,见图4)。
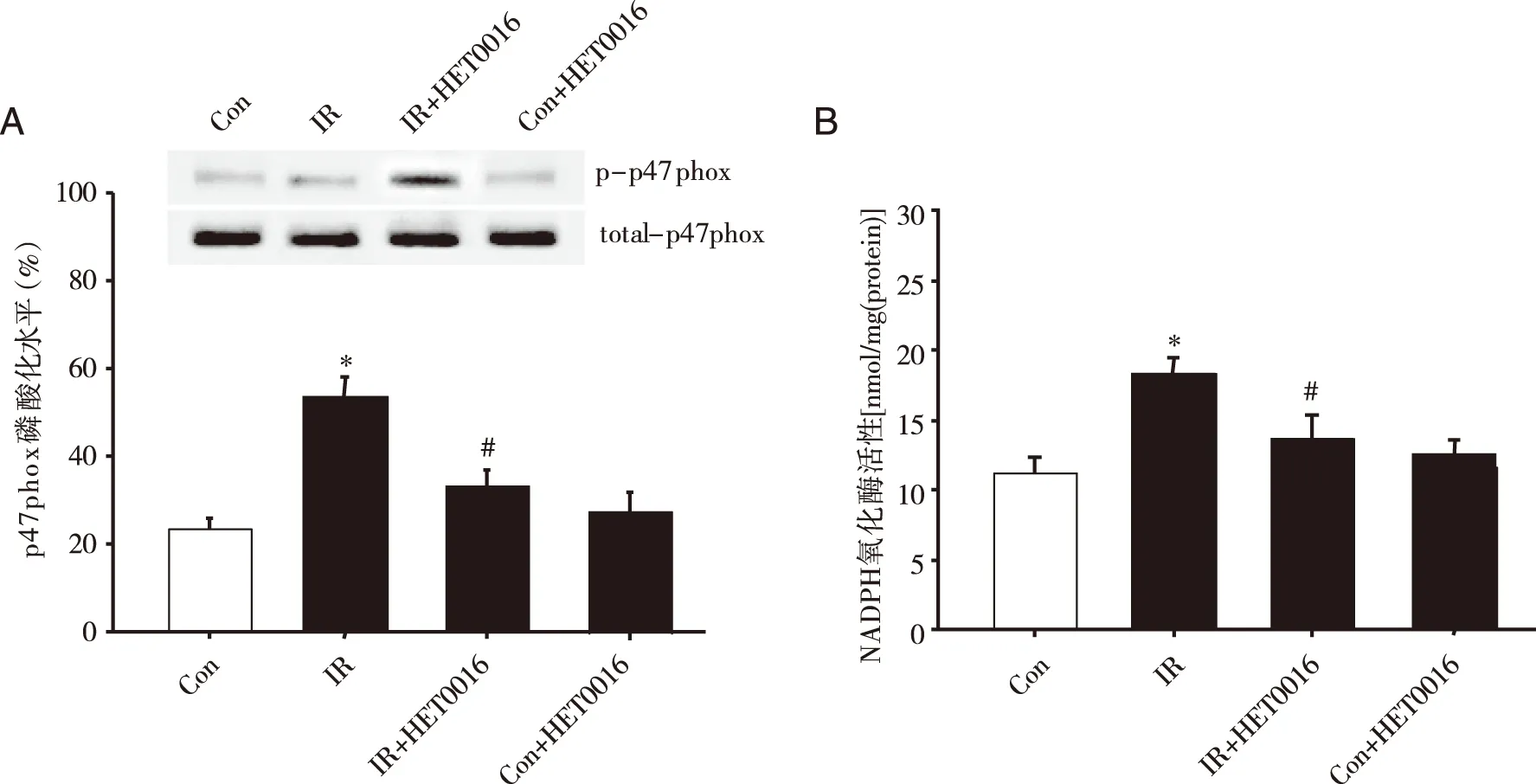
A:p47phox亚基磷酸化免疫印迹结果和p47phox亚基磷酸化统计分析;B:NADPH氧化酶活性统计分析;*:P<0.05,与Con组相比;#:P<0.05,与IR组相比。图4 HET0016对NADPH氧化酶亚基p47phox磷酸化及全酶活性的影响
3 讨论
近年来一些研究报道了在心肌缺血再灌注损伤(myocardial ischemia reperfusion injury,MIRI)中,冠状静脉血浆内20-HETE水平含量增高,同时心肌组织中CYP ω-羟化酶活性显著增强[4-5]。在大鼠离体心脏灌流中应用非特异性CYP450酶的抑制剂(如氯霉素、西米替丁、磺胺苯吡唑等),或者在缺血再灌注中应用特异性CYP ω-羟化酶抑制剂(如17ODYA、DDMS)均可减少心肌再灌注后的损伤,与之相反,当外源加入20-HETE后,可加重缺血再灌注损伤,降低了心肌再灌注后功能的恢复[4-5,8]。如上发现与本研究中20-HETE对离体缺血再灌注后的心脏功能影响结果相一致,本研究发现当应用20-HETE合成酶的特异性阻断剂HET0016后,减少MIRI进程中20-HETE合成可明显改善再灌注后心肌力学指标,与IR组心脏相比较,IR+HET0016组心脏再灌注后心肌功能显著恢复,再灌注结束时左心室发展压(LVDP)、左心室内压最大上升/下降速率(±dP/dtmax)出现明显回升,而左心室末期舒张压(LVEDP)明显下降;同时心肌梗死面积显著减少(P<0.05)。这些数据表明20-HETE参与到MIRI当中,但是其加重再灌注后损伤、降低心肌功能的分子细胞机制尚需进一步探究。
众多研究证明,ROS的过量生成在MIRI进程中发挥了主要作用,使用抗氧化剂清除ROS可明显改善心肌再灌注后功能的恢复,被认为是引起缺血再灌注损伤的主要启动因子[6-7]。由于心脏中缺乏抗氧化酶,因此对于ROS的敏感性高于其他组织器官。一般认为ROS从再灌注开始便产生,4~7 min后达到峰值,当心肌细胞内产生过量ROS后,可以损伤亚细胞组分,如降解膜磷脂,使蛋白质过氧化损伤及攻击DNA。这其中过量生成的ROS对细胞内可收缩性蛋白的损伤可能直接导致了心肌收缩性能的下降。已有研究表明,心肌缺血后的肌钙蛋白及肌动蛋白均发生了氧化损伤而影响缺血后的心肌的收缩功能[9-10]。这提示20-HETE造成心肌缺血再灌注后心肌血流动力学下降可能与其促进ROS生成增多、造成收缩相关蛋白氧化损伤有关。因此,本研究应用DHE荧光探针法通过激光共聚焦显微镜,检测了在MIRI进程中抑制20-HETE的合成后对离体心肌再灌后ROS生成的影响,发现缺血再灌注诱导心肌组织内ROS大量的生成,在此基础上加入HET0016处理后,心肌组织中的ROS荧光强度显著减少(P<0.05)。如上结果表明,在MIRI中,20-HETE诱发了心肌组织过量ROS的生成。
缺血再灌注中ROS生成主要来源于线粒体呼吸链、NADPH氧化酶、一氧化氮合酶等。在这些来源中,NADPH氧化酶不同于其他来源的复杂之处在于其生成ROS受到高度的调控。NADPH氧化酶由多个亚基构成,包括位于胞膜上的gp91phox和p22phox结合而成的二聚体,另外包括位于胞质内的p47phox、p40phox、p67phox以及Rac2等亚基成分[11],这其中p47phox亚基是NADPH氧化酶胞质和胞膜各组分之间的连接蛋白,首先在一些刺激因素作用下,p47phox被磷酸化,进而与p67phox发生相互作用,由胞质转位到胞膜上而激活NADPH氧化酶产生ROS,因此p47phox磷酸化是NADPH氧化酶激活的关键步骤[12-13]。目前有研究报道MIRI进程中NADPH氧化酶的激活可诱导产生过量的ROS而造成心肌过氧化损伤[11,14-15]。因此,为了探究在心肌细胞MIRI进程中,20-HETE诱导的ROS生成是否与其激活NADPH氧化酶相关,本研究检测了MIRI中抑制20-HETE合成后对NADPH氧化酶活性以及p47phox亚基磷酸化作用的影响。通过研究发现,IR+HET0016组心肌组织中磷酸化的p47phox亚基显著低于IR组(P<0.05)。通过对NADPH氧化酶全酶活性检测也发现,再灌注后IR+HET0016组心肌组织NADPH氧化酶活性比IR组明显降低(P<0.05)。如上结果表明,在MIRI进程中,20-HETE通过激活NADPH氧化酶的活性,从而诱导过量ROS的生成,最终加重心肌再灌注后的损伤。
综上,本研究发现在大鼠离体缺血再灌注模型中,导致所述MIRI的机制与20-HETE提高p47phox磷酸化水平从而激活NADPH氧化酶,诱导过量ROS的生成有关,因此造成心肌功能下降,梗死面积增加,为临床治疗缺血性心肌病提供了一定的理论依据。
[1] Edson K Z, Rettie A E. CYP4 enzymes as potential drug targets: focus on enzyme multiplicity, inducers and inhibitors, and therapeutic modulation of 20-hydroxyeicosatetraenoic acid (20-HETE) synthase and fatty acid omega-hydroxylase activities[J]. Curr Top Med Chem, 2013, 13(12): 1429-1440.
[2] Ding Y, Wu C C, Garcia V, et al. 20-HETE induces remodeling of renal resistance arteries independent of blood pressure elevation in hypertension[J]. Am J Physiol Renal Physiol, 2013, 305(5): 753-763.
[3] Roman R J,Akbulut T,Park F, et al.20-HETE in acute kidney injury[J].Kidney Int,2011,79(1):10-13.
[4] Ishihara Y, Sekine M, Nakazawa M, et al. Suppression of myocardial ischemia-reperfusion injury by inhibitors of cytochrome P450 in rats[J]. Eur J Pharmacol, 2009, 611(1-3): 64-71.
[5] Nithipatikom K, Endsley M P, Moore J M, et al. Effects of selective inhibition of cytochrome P-450 omega-hydroxylases and ischemic preconditioning in myocardial protection[J]. Am J Physiol Heart Circ Physiol, 2006, 290(2): 500-505.
[6] Schriewer J M, Peek C B, Bass J, et al. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion[J]. J Am Heart Assoc, 2013, 2(2): e000159.
[7] Wang X, Jian C, Zhang X, et al. Superoxide flashes: elemental events of mitochondrial ROS signaling in the heart [J]. J Mol Cell Cardiol, 2012, 52(5): 940-948.
[8] Granville D J, Tashakkor B, Takeuchi C, et al. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors[J]. Proc Natl Acad Sci U S A, 2004, 101(5): 1321-1326.
[9] Murphy A M, Kogler H, Georgakopoulos D, et al. Transgenic mouse model of stunned myocardium[J]. Science, 2000, 287(5452): 488-491.
[10] Powell S R, Gurzenda E M, Wahezi S E. Actin is oxidized during myocardial ischemia[J]. Free Radic Biol Med, 2001, 30(10): 1171-1176.
[11] Rodino-Janeiro B K, Paradela-Dobarro B, Castineiras-Landeira M I, et al. Current status of NADPH oxidase research in cardiovascular pharmacology[J]. Vasc Health Risk Manag, 2013, 9(1): 401-428.
[12] El-Benna J, Dang P M, Gougerot-Pocidalo M A, et al. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases[J]. Exp Mol Med, 2009, 41(4): 217-225.
[13] Belambri S A, Hurtado-Nedelec M, Senator A, et al. Phosphorylation of p47phox is required for receptor-mediated NADPH oxidase/NOX2 activation in Epstein-Barr virus-transformed human B lymphocytes[J]. Am J Blood Res, 2012, 2(3): 187-193.
[14] Qin F, Simeone M, Patel R. Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction[J]. Free Radic Biol Med, 2007, 43(2): 271-281.
[15] Yu Q, Lee C F, Wang W, et al. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury[J]. J Am Heart Assoc, 2014, 3(1): 555.
[收稿2016-05-08;修回2016-06-16]
(编辑:王静)
Effect of 20-HETE on NADPH oxidase activity and ROS production of rats with myocardial ischemia reperfusion injury
HanYong1,GuoLirong2,HeYan1,LiuWanli1,WangMei1,ZhaoHongyan1,HeYa1
(1. Department of Physiology, School of Basic Medical Sciences, Zunyi Medical University, Zunyi Guizhou 563099, China; 2. Morphological Laboratory, School of Basic Medical Sciences, Zunyi Medical University, Zunyi Guizhou 563099, China)
Objective To investigate the effect and mechanism of 20-HETE aggravates myocardial ischemia reperfusion injury in rats.Methods Male Wistar rats were randomly divided into normal control group (Con), ischemia-reperfusion group (IR), IR+inhibitor of 20-HETE production (HET0016) group and Con+HET0016 group, with 12 rats in each group. Experiments were performed in isolated rat hearts subjected to 35 min of ischemia followed by 40 min of reperfusion in Langendorff preparations. Cardiac contractility was continuously recorded with the Powerlab/8P system. Myocardial infarct size was measured by TTC staining. The level of reactive oxygen species (ROS) was determined by DHE fuorescence. Phosphorylation of p47phox, a cytosolic component of NADPH oxidase, was detected by Western blot method. NADPH oxidase activity was evaluated by NADPH-dependent superoxide production examined using SOD-inhibitable cytochrome c reduction.Results At the end of reperfusion, administration of HET0016 (an inhibitor of 20-HETE production, 1 μmol/l) significantly ameliorated the inhibited cardiac function induced by IR, including augmented LVDP from (48.6±3.4)% to (71.7±3.5)%, +dP/dtmaxfrom (51.9±2.1)% to (69±3.2)% and -dP/dtmaxfrom (47.1±3.6)% to (64.1±3.8)% (P<0.05). Treatment of the heart with HET0016 significantly attenuated the IR-induced increase in cardiac infarct size by 37.2% (P<0.05). Compared with the IR group, IR+HET0016 treatment significantly reduced the IR-induced increase in DHE fluorescent intensity by 45.8% (P<0.05). Finally, HET0016 also decreased the phosphorylation level of p47phoxand depressed NADPH oxidase activity resepectively(P<0.05).Conclusion 20-HETE could stimulate NADPH oxidase-derived superoxide production, which aggravated IR-induced myocardial injury.
20-HETE; rat; myocardial ischemia reperfusion injury; reactive oxygen species; NADPH oxidase
国家自然科学基金资助项目(NO:81460040);贵州省科学技术基金资助项目(NO:黔科合LH字[2014]7544)。
R542.2
A
1000-2715(2016)04-0366-06
猜你喜欢
麦类作物学报(2022年3期)2022-05-19
临床与实验病理学杂志(2022年3期)2022-04-06
昆明医科大学学报(2020年12期)2021-01-26
食品与生物技术学报(2021年4期)2021-01-17
世界最新医学信息文摘(2020年68期)2020-12-25
烟台大学学报(自然科学与工程版)(2020年1期)2020-02-08
中成药(2019年12期)2020-01-04
天然产物研究与开发(2018年8期)2018-09-10
奥秘(2016年10期)2016-12-17
园艺与种苗(2015年10期)2015-02-27
