
坏死性凋亡介导高糖引起的人脐静脉内皮细胞损伤*
2016-10-26 05:34:38林佳琼陈美姬郭瑞鲜张伟杰智喜梅邓海鸥黎映兰
中国病理生理杂志 2016年9期
林佳琼, 陈美姬, 郭瑞鲜, 张伟杰, 智喜梅, 邓海鸥, 许 翎, 黎映兰, 吴 文△
(1南方医科大学,广东 广州 510515; 2广东省人民医院, 广东省医学科学院,广东省老年医学研究所东病区内分泌科,广东 广州 510080; 3中山大学附属医院第一医院黄埔院区儿科,广东 广州 510700;4中山大学中山医学院生理学教研室,广东 广州 510080)
坏死性凋亡介导高糖引起的人脐静脉内皮细胞损伤*
林佳琼1, 2, 陈美姬3, 郭瑞鲜4, 张伟杰2, 智喜梅2, 邓海鸥2, 许 翎2, 黎映兰2, 吴 文2△
(1南方医科大学,广东 广州 510515;2广东省人民医院, 广东省医学科学院,广东省老年医学研究所东病区内分泌科,广东 广州 510080;3中山大学附属医院第一医院黄埔院区儿科,广东 广州 510700;4中山大学中山医学院生理学教研室,广东 广州 510080)
目的: 研究坏死性凋亡是否介导高糖(HG)诱导的人脐静脉内皮细胞(human umbilical vein endothelial cells,HUVECs)损伤。方法: CCK-8法检测细胞存活率;Western blot法测定受体相互作用蛋白3(RIP3)、cleaved caspase-3的蛋白水平;罗丹明123染色荧光显微镜照相法检测线粒体膜电位(mitochondrial membrane potential,MMP);双氯荧光素(DCFH-DA)染色荧光显微镜照相法测定胞内活性氧簇(reactive oxygen species,ROS)的水平。结果: 应用不同浓度葡萄糖(10、20和40 mmol/L)处理HUVECs 24 h,RIP3的蛋白水平随葡萄糖剂量增加而升高,40 mmol/L时达高峰;应用40 mmol/L葡萄糖处理HUVECs 3 h、6 h、9 h、12 h和24 h能上调RIP3的蛋白水平,于9 h达最高峰;应用20 μmol/L凋亡蛋白酶抑制剂Z-VAD-FMK预处理HUVECs 30 min促进RIP3表达;应用100 μmol/L坏死性凋亡抑制剂necrostatin-1预处理HUVECs 1 h能抑制HG诱导HUVECs的细胞存活率降低,ROS过度生成及MMP丢失,但能升高 cleaved caspase-3的蛋白水平。结论: 坏死性凋亡介导高糖引起的人脐静脉内皮细胞损伤,但与内皮细胞凋亡存在负相关。
坏死性凋亡; 细胞凋亡; 高糖; 人脐静脉内皮细胞; Necrostatin-1
高血糖可通过多种病理生理机制引起血管内皮细胞功能失调。血管内皮功能异常主要是内皮细胞的损伤或死亡导致功能的紊乱,从而导致多种并发症的发生。传统认为细胞死亡方式包括凋亡、坏死及自噬。既往认为凋亡是caspase依赖的可调控的程序性细胞死亡[1],而坏死是一种被动的非可控的细胞死亡方式。然而,近年来,Degterev等[2]报道了一种新的细胞死亡方式,一种与坏死具有相似的形态学特征,但其方式为可调控的非caspase依赖性的程序性细胞坏死,即在caspase抑制的条件下,死亡受体与配体的结合可触发坏死性凋亡(necroptosis)。受体交互作用蛋白1(receptor-interacting protein 1,RIP1)和受体交互作用蛋白3(receptor-interacting protein 3,RIP3)形成复合物和磷酸化是坏死性凋亡发生的关键,因此是坏死性凋亡发生的特异性生化标志物[3-4]。我们的研究证实[5]高糖可引起人脐静脉血管内皮细胞的损伤,包括细胞存活率降低、活性氧簇(reactive oxygen species,ROS)生成增多、线粒体损伤及细胞凋亡增加,近年,坏死性凋亡被认为是可能参与高血糖引起心肌死亡的一个重要的介导者[6],因此,我们推测高糖也可以通过坏死性凋亡引起血管内皮细胞的损伤。
为此,本文在人脐静脉内皮细胞(human umbilical vein endothelial cells,HUVECs)建立高糖损伤模型,旨在探讨:高糖是否引起HUVECs产生坏死性凋亡;坏死性凋亡与凋亡之间的相互作用;坏死性凋亡是否参与高糖诱导HUVECs的多种损伤。
材 料 和 方 法
1 材料
坏死性凋亡抑制剂necrostatin-1(Nec-1)、凋亡蛋白酶抑制剂Z-VAD-FMK、DCFH-DA和罗丹明123(rhodamine 123,Rh123)购自Sigma;CCK-8试剂盒购自Dojindo;胎牛血清(fetal bovine serum,FBS)购自Gibco;cleaved caspase-3抗体购自Cell Signaling Technology;RIP3抗体购自Abcam;HUVECs购自广州吉妮欧公司。
2 方法
2.1 人脐静脉内皮细胞的体外培养 细胞培养在含10%胎牛血清的低糖DMEM培养基,同时加入1×105U/L青霉素和100 mg/L链霉素,置于5% CO2、37 ℃的培养箱中培养。待细胞融合达80%后,用0.25%胰蛋白酶-EDTA消化,适度消化后加完全培养基终止胰蛋白酶的消化作用。用灭菌Tip头将瓶壁细胞吹落形成细胞悬液。1 200 r/min离心5 min, 弃上清, 按1∶3传代。实验前换用无血清的DMEM培养液培养12 h,然后进行分组实验。
2.2 实验分组 实验分为4组:正常对照(control)组;高糖(high glucose, HG)损伤组:应用40 mmol/L葡萄糖处理HUVECs 24 h;Z-VAD-FMK+HG组:20 μmol/L Z-VAD-FMK 作用于HUVECs 30 min,撤去,用PBS洗2次,接着用40 mmol/L葡萄糖作用 24 h;Nec-1+HG组:100 μmol/L Nec-1 作用于HUVECs 1 h,撤去,用PBS洗2次,接着用40 mmol/L葡萄糖作用24 h;Z-VAD-FMK组:20 μmol/L Z-VAD-FMK作用于HUVECs 30 min,撤去,用PBS洗2次,无血清培养基DMEM培养24 h;Nec-1组:100 μmol/L Nec-1作用于HUVECs 1 h,撤去,用PBS洗2次,无血清培养基DMEM培养24 h。
2.3 CCK-8法测定细胞存活率 根据CCK-8试剂盒说明书检测HUVECs活力。将HUVECs以每孔5 000细胞的密度接种于96孔板,当细胞生长80%时,给予不同处理。于每孔加入无血清培养基稀释的CCK-8工作液100 μL,轻摇,37 ℃孵育2 h,使用酶标仪检测各孔吸光度(A),波长设定为450 nm。按公式:细胞存活率(%)=处理组A/对照组A×100%,求出处理组细胞存活率,重复5次。
2.4 Western blot法检测RIP3和cleaved caspase-3的蛋白水平 将HUVECs接种于60 mm培养皿中,贴壁生长至80%时给予不同处理因素,弃培养液,用预冷的PBS洗2次,各皿加入80 μL裂解液后置4 ℃裂解30 min,12 000 r/min离心15 min,取上清,蛋白浓度采用BCA法进行测定。总蛋白经SDS-PAGE分离后,转移到PVDF膜上。用5%脱脂奶粉封闭90 min,分别加入抗RIP3和cleaved caspase-3的I抗(1∶1 000)4 ℃过夜,然后用TBST洗10 min 3次,加入II抗(1∶10 000),室温孵育60 min,然后再用TBST洗10 min 3次。PVDF膜用ECL发光液显色,暗室曝光,凝胶成像系统扫描分析结果。
2.5 细胞内ROS含量的检测 2’-7’-二氯荧光乙酰乙酸盐(DCFH-DA)是一种自身不能发荧光的化合物,可自由穿过细胞膜,被胞内活性氧氧化为有荧光的DCF。通过检测DCF的荧光强度即可反映细胞内ROS的水平。将HUVECs接种于6孔培养板中,当细胞生长贴壁生长至80%时,给予各实验组不同的处理因素后,弃培养液,PBS 冲洗3次,将HUVECs与10 μmol/L DCFH-DA于37 ℃孵育30 min,再用PBS冲洗3次。在荧光显微镜下随机选取5个不重复区摄片,采用ImageJ 1.41软件分析5个视野绿色荧光强度平均值,进而对每组的各个样本进行统计分析。实验重复5次。
2.6 细胞线粒体膜电位(mitochondria membrane potential,MMP)的测定 Rh123是一种可被线粒体吸收的绿色荧光染料,其吸收值随细胞MMP的改变而变化,因此可根据其荧光强度来间接反映MMP的高低。将HUVECs接种于6孔培养板中,贴壁生长至大约80%时,给予各实验组不同的处理因素,弃培养液,用PBS冲洗3次,予10 μg/L Rh123于37 ℃避光孵育45 min, 再用PBS冲洗3次。在荧光显微镜下随机地选取5个不重复区摄片。用ImageJ 1.41软件分析各视野平均荧光强度,进而对每组的各个样本进行统计分析。实验重复5次。
3 统计学处理
实验数据以均数±标准差(mean±SD)表示,用SPSS 20.0统计学软件进行统计分析,组间比较采用单因素方差分析(one-way ANOVA),采用SNK-q检验进行均数之间的两两比较,以P<0.05为差异有统计学意义。
结 果
1 坏死性凋亡抑制剂Nec-1对抗高糖引起的细胞毒性
予HG(40 mmol/L葡萄糖)处理HUVECs 24 h,可引起细胞毒性,使细胞存活率降低,与对照组相比,差异具有统计学显著性(P<0.01)。在高糖处理HUVECs前,予100 μmol/L Nec-1预处理HUVECs 1 h,可明显抑制HG的细胞毒性,使细胞存活率明显上升,与HG组相比,差异有统计学显著性(P<0.01)。而单独100 μmol/L Nec-1预处理HUVECs 1 h不引起细胞毒性,见图1。
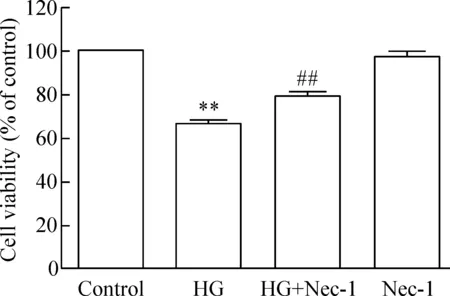
Figure 1.Inhibitor of necroptosis Nec-1 protected HUVECs against HG-induced cytotoxicity. Mean±SD.n=5.**P<0.01vscontrol;##P<0.01vsHG.
图1 坏死性凋亡抑制剂Nec-1保护HUVECs对抗高糖引起的细胞毒性
2 高糖促进人脐静脉内皮细胞的RIP3蛋白表达
使用不同浓度(10~40 mmol/L)的葡萄糖处理HUVECs 24 h均能促进RIP3蛋白的表达,与对照组分别相比,差异均有统计学显著性(P<0.01),且葡萄糖在40 mmol/L时RIP3的表达最高;使用HG(40 mmol/L)处理HUVECs 3~24 h可上调RIP3蛋白水平(P<0.01),其中在9 h时RIP3的表达水平达到最高峰,见图2。
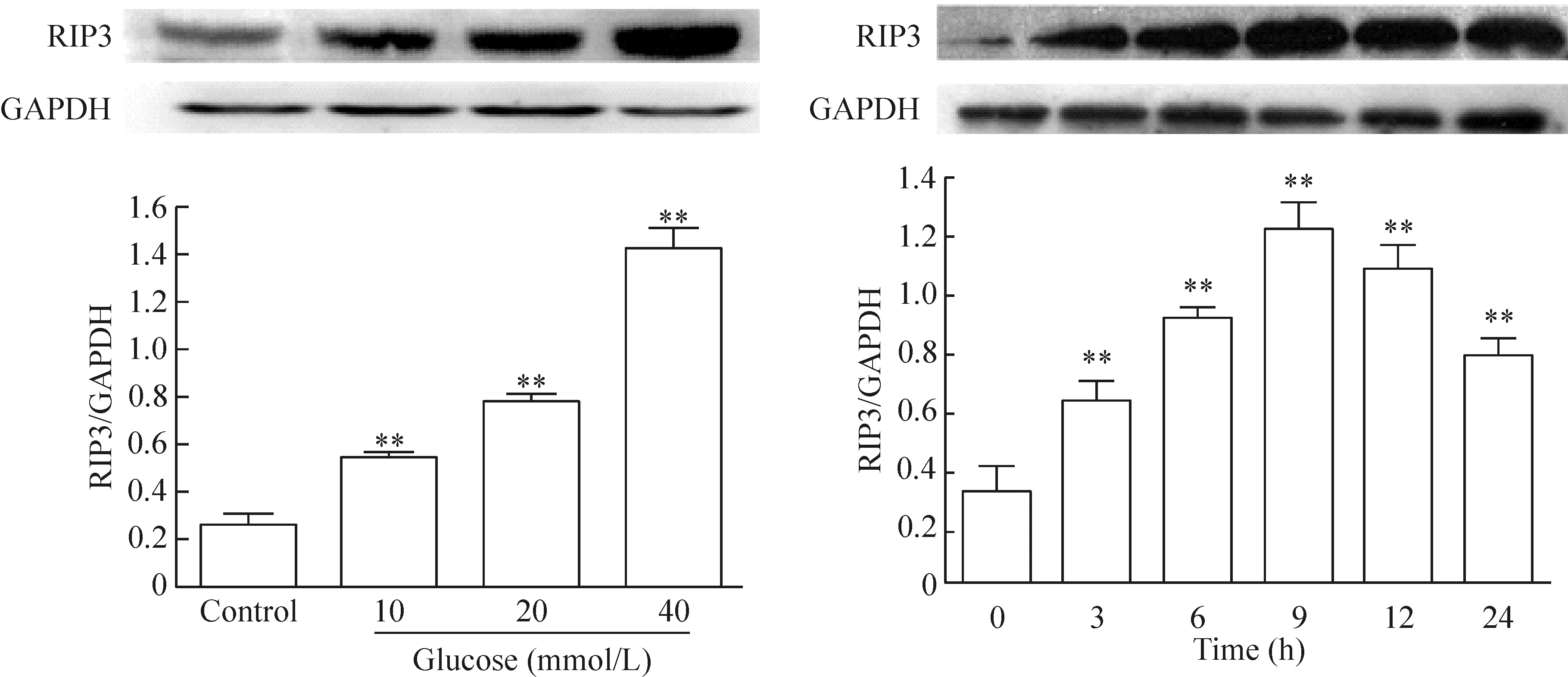
Figure 2. High glucose (40 mmol/L glucose, HG) up-regulated the expression level of RIP3 in the HUVECs. Mean±SD.n=3.**P<0.01vscontrol.
图2 高糖促进HUVECs的RIP3表达
3 坏死性凋亡介导高糖引起的人脐静脉内皮细胞的氧化应激
如图3显示,HG处理HUVECs 24 h可使胞内DCFH的平均荧光强度(MFI)明显增强,与正常对照组相比,差异有统计学显著性(P<0.01)。应用100 μmol/L浓度的Nec-1预处理1 h可使MFI从(163.2±18.5)%减少至(122.0±36.4)%,可明显抑制HG诱导的胞内ROS堆积,与HG组相比,差异有统计学显著性(P<0.01)。而应用100 μmol/L浓度的Nec-1处理HUVECs 1 h 对胞内ROS生成无明显作用。
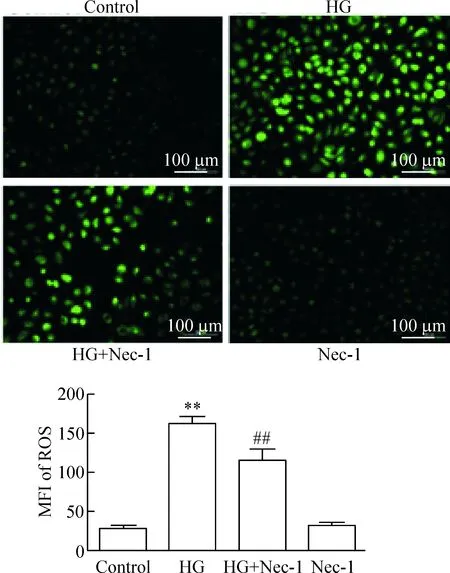
Figure 3. The inhibitor of necroptosis Nec-1 reduced HG-induced accumulation of reactive oxygen species (ROS) in the HUVECs. Mean±SD.n=5.**P<0.01vscontrol;##P<0.01vsHG.
图3 坏死性凋亡通路抑制剂Nec-1减少高糖引起的HUVECs内活性氧的堆积
4 坏死性凋亡介导高糖引起的人脐静脉内皮细胞的线粒体损伤
HG作用HUVECs 24 h使胞内Rh123的MFI明显降低,从(258.2±32.5)%降低至(119.2±9.4)%,两组比较差异有统计学显著性(P<0.01)。但是,在HG 处理HUVECs前,应用100 μmol/L浓度的Nec-1预处理1 h可使MFI升高至(169.8±13.4)%,与HG处理组相比,差异有统计学显著性(P<0.01)。100 μmol/L浓度的Nec-1作用HUVECs 1 h对胞内MMP生成无明显作用,见图4。
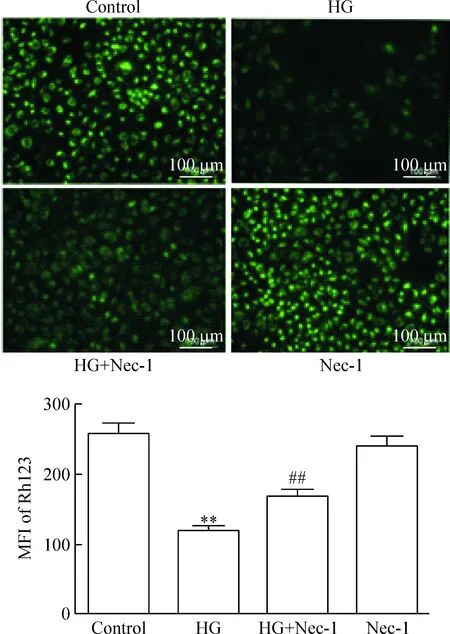
Figure 4. The inhibitor of necroptosis Nec-1 attenuated HG-induced dissipation of mitochondrial membrance potential (MMP) in the HUVECs. Mean±SD.n=5.**P<0.01vscontrol;##P<0.01vsHG.
图4 坏死性凋亡通路抑制剂Nec-1减少高糖引起的HUVECs线粒体膜电位丢失
5 坏死性凋亡抑制剂促进HG引起的人脐静脉内皮细胞的凋亡
使用HG处理HUVECs 24 h, cleaved caspase-3的蛋白水平明显增加,与对照组相比,差异有统计学显著性(P<0.01);值得注意的是,在HG处理HUVECs前,应用100 μmol/L浓度的坏死性凋亡抑制剂Nec-1预处理HUVECs 1 h,能明显促进cleaved caspase-3蛋白水平的上调,与HG处理组比较,差异有统计学显著性(P<0.01)。100 μmol/L浓度的Nec-1本身对cleaved caspase-3的基础水平无明显影响,见图5。
6 凋亡抑制剂促进HG引起的人脐静脉内皮细胞的坏死性凋亡
HG处理HUVECs 24 h,RIP3蛋白表达明显增加,与对照组相比,差异有统计学显著性(P<0.01)。在HG处理HUVECs前,应用20 μmol/L凋亡蛋白酶抑制剂Z-VAD-FMK预处理HUVECs 30 min明显促进RIP3蛋白的表达,与HG处理组比较,差异有统计学显著性(P<0.05)。20 μmol/L浓度的Z-VAD-FMK本身对RIP3的基础表达水平无明显影响,见图6。
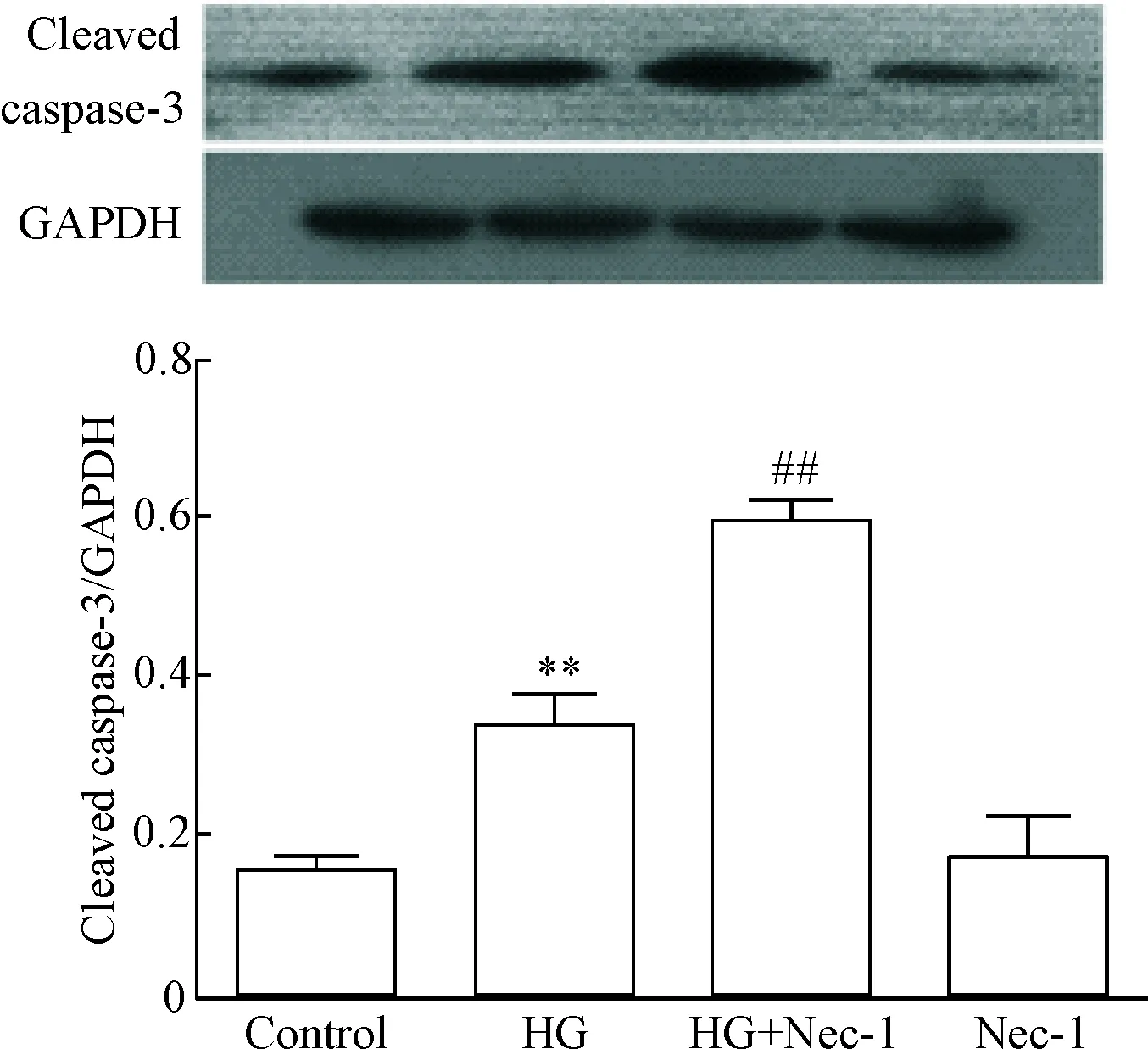
Figure 5.The inhibitor of necroptosis Nec-1 up-regulated the protein level of cleaved caspase-3 in the HUVECs. Mean±SD.n=3.**P<0.01vscontrol;##P<0.01vsHG.
图5 坏死性凋亡抑制剂Nec-1促进HUVECs的cleaved caspase-3蛋白水平上调
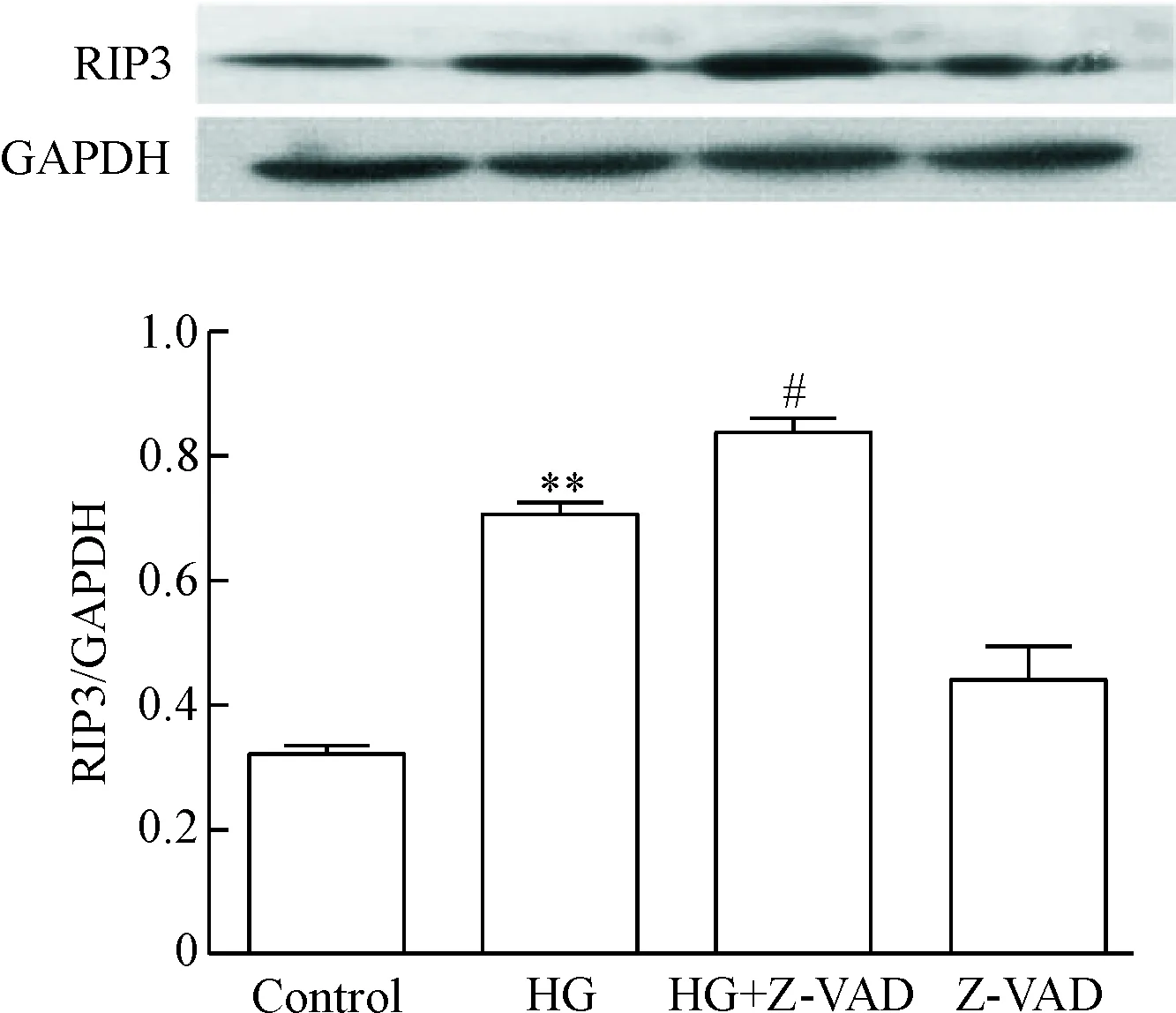
Figure 6.The inhibitor of apoptosis Z-VAD-FMK enhanced the expression level of RIP3 in the HUVECs. Mean±SD.n=3.**P<0.01vscontrol;#P<0.05vsHG.
图6 凋亡蛋白酶抑制剂Z-VAD-FMK促进RIP3蛋白的表达
讨 论
既往研究认为,坏死是一种意外死亡,与凋亡程序性死亡截然不同,它是无序的被动死亡,无从调控。近年,坏死性凋亡的发现颠覆了传统的认识,它将为临床疾病的治疗提供新的靶点。坏死性凋亡形态上既具有坏死的特性(包膜完整性破坏、细胞体积及胞内细胞器膨胀),可以通过释放损伤相关分子模式(damage-associated molecular patterns,DAMPs)和激活炎症小体NLRP3而促进炎症的发生[7],又具备有凋亡通路可调控死亡的程序性。越来越多的研究表明坏死性凋亡在多种疾病和组织损伤如脑缺血、肾损伤及心肌梗死中起着重要作用[8],Luedde 等[9]报道,在缺血心肌损伤中,RIP3表达增多;敲除RIP3则能保护心肌对抗缺血性损伤。也有研究发现[10],坏死性凋亡特别是RIP3与动脉粥样硬化的发展有关。近年,高血糖对坏死性凋亡的作用引起学者们的重视。在链脲霉素诱导的糖尿病小鼠发生心肌纤维化和心肌肥厚的同时,伴随着心肌RIP3表达增多[11]。但是,高糖对血管内皮细胞RIP3的影响迄今未明。为了证实坏死性凋亡是否与高糖引起的血管病变有关,我们进行了多方面的探讨。本研究观察到,随着葡萄糖浓度的增加,HUVECs的RIP3蛋白表达明显增加,提示HG是诱导血管内皮细胞坏死性凋亡的诱导剂。进一步研究还发现,坏死性凋亡抑制剂Nec-1可以抑制HG引起的血管内皮细胞损伤,使细胞存活率升高,ROS生成及MMP丢失均减少。提示HG可以通过坏死性凋亡诱导人脐静脉内皮细胞损伤。
许多研究发现,RIP1和RIP3形成复合物和磷酸化是坏死性凋亡发生的关键[12-13],RIP复合物处在凋亡与坏死性凋亡的分叉口,起着决定细胞死亡方式的调控作用。因此,凋亡与坏死性凋亡存在密切的联系,二者存在部分的重合。坏死性凋亡抑制剂Nec-1可以特异性地抑制坏死性凋亡,而不影响正常细胞的生理功能,可明确细胞是否发生坏死性凋亡。而Caspase抑制剂Z-VAD-FMK是一种泛aspase抑制剂,可以抑制由caspase激活引起的细胞凋亡。对于凋亡与坏死性凋亡的相互转化,目前研究主要集中于两个方面。一方面是抑制凋亡后,可以促进坏死性凋亡的发生[3-4]。本研究也观察到了这一点,当给予凋亡蛋白酶抑制剂Z-VAD抑制凋亡时,RIP3的表达明显升高。另一方面,抑制坏死性凋亡可以抑制或促进凋亡发生。有研究表明,坏死性凋亡抑制剂Nec-1可以降低cleaved caspase-3的蛋白水平[14]。而相反地,Han等[15]发现,高浓度紫草素可激活癌细胞株HL60的坏死性凋亡通路使细胞死亡,而在使用Nec-1抑制坏死性凋亡后,细胞转而发生凋亡。我们在高糖损伤HUVECs模型中,发现应用Nec-1抑制坏死性凋亡后,cleaved caspase-3的蛋白水平明显上调,提示促进细胞转向凋亡,这与Han等[15]的结果近似。上述这些不同的研究结论提示,在不同的病理损伤及不同的细胞实验模型中,细胞死亡方式的相互转化形式可能不尽一致,很值得进一步探讨。
综上所述,本研究证实:(1) HG可诱导HUVECs发生坏死性凋亡;(2) 坏死性凋亡介导HG引起的氧化应激与线粒体损伤;(3) 坏死性凋亡与凋亡可存在相互转化,即抑制坏死性凋亡后,细胞死亡可转化为凋亡,反之亦然。上述结果对进一步揭示糖尿病血管病变病理生理机制有重要的意义。
[1] Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematodeC.elegans[J]. Cell, 1986, 44(6):817-829.
[2] Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury[J]. Nat Chem Biol, 2005, 1(2):112-119.
[3] Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation[J]. Cell, 2009, 137(6):1112-1123.
[4] He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha[J]. Cell, 2009, 137(6):1100-1111.
[5] 廖静秋,林佳琼,张伟杰,等. JAK/STAT信号通路在高糖诱导人脐静脉内皮细胞损伤中的作用[J]. 中国病理生理杂志, 2016, 32(3):392-397.
[6] Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart[J]. Circ Res, 2011, 108(8):1017-1036.
[7] Lawlor KE, Khan N, Mildenhall A, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL[J]. Nat Commun, 2015, 6:6282.
[8] Linkermann A, Green DR. Necroptosis[J]. N Engl J Med, 2014, 370(5):455-465.
[9] Luedde M, Lutz M, Carter N, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction[J]. Cardiovasc Res, 2014, 103(2):206-216.
[10]Lin J, Li H, Yang M, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development[J]. Cell Rep, 2013, 3(1):200-210.
[11]Liu YS, Huang ZW, Wang L, et al. Sitagliptin alleviated myocardial remodeling of the left ventricle and improved cardiac diastolic dysfunction in diabetic rats[J]. J Pharmacol Sci, 2015, 127(3):260-274.
[12]Degterev A, Zhou W, Maki JL, et al. Assays for necroptosis and activity of RIP kinases[J]. Methods Enzymol, 2014, 545:1-33.
[13]Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis[J]. Science, 2009, 325(5938):332-336.
[14]Zhang N, Chen Y, Jiang R, et al. PARP and RIP 1 are required for autophagy induced by 11’-deoxyverticillin A, which precedes caspase-dependent apoptosis[J]. Autophagy, 2011, 7(6):598-612.
[15]Han W, Xie J, Li L, et al. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis[J]. Apoptosis, 2009, 14(5):674-686.
(责任编辑: 林白霜, 罗 森)
Necroptosis mediates high glucose-induced injury in human umbilical vein endothelial cells
LIN Jia-qiong1, 2, CHEN Mei-ji3, GUO Rui-xian4, ZHANG Wei-jie2, ZHI Xi-mei2, DENG Hai-ou2, XU Ling2, LI Ying-lan2, WU Wen2
(1SouthernMedicalUniversity,Guangzhou510515,China;2DepartmentofEndocrinology,EastWard,GuangdongGeriatricInstitute,GuangdongAcademyofMedicalSciences,GuangdongGeneralHospital,Guangzhou510080,China;3DepartmentofPediatrics,HuangpuDivisionofTheFirstAffiliatedHospitalofSunYat-senUniversity,Guangzhou510700,China;4DepartmentofPhysiology,ZhongshanSchoolofMedicine,SunYat-senUniversity,Guangzhou510080,China.E-mail:wuwen1964@163.com)
AIM: To explore whether necroptosis contributes to the high glucose (HG)-induced damage in human umbilical vein endothelial cells (HUVECs). METHODS: The protein levels of receptor-interacting protein 3 (RIP3) and cleaved caspase-3 were detected by Western blot. The intracellular levels of reactive oxygen species (ROS) were determined by DCFH-DA staining followed by photofluorography. Mitochondrial membrane potential (MMP) was measured by rhodamine 123 staining followed by photofluorography. RESULTS: Treatment of HUVECs with HG at different concentrations (10, 20 and 40 mmol/L glucose) for 24 h gradually enhanced the expression levels of RIP3. Treatment of HUVECs with HG (40 mmol/L glucose) for different time (3 h, 6 h, 9 h, 12 h and 24 h) also up-regulated the expression levels of RIP3, peaking at 9 h. Pretreatment of HUVECs with 20 μmol/L Z-VAD-FMK (an inhibitor of caspase) for 30 min before exposure to HG enhanced the expression level of RIP3. Pretreatment of HUVECs with 100 μmol/L necrostatin-1 (an inhi-bitor of necroptosis) for 1 h before exposure to HG alleviated the HG-induced injuries, such as a decrease in cell viability, an increase in ROS generation and dissipation of MMP, but up-regulated the protein level of cleaved caspase-3. CONCLUSION: Necroptosis mediates HG-induced injury in HUVECs. There is a negative interacting between necroptosis and apoptosis.
Necroptosis; Apoptosis; High glucose; HUVECs; Necrostatin-1
杂志网址: http://www.cjpp.net
1000- 4718(2016)09- 1608- 06
2016- 04- 25
2016- 05- 31
国家自然科学基金资助项目(No. 81450062);广东省自然科学基金资助项目(No. 2015A030313872)
△通讯作者 Tel: 020-83827812; E-mail: wuwen1964@163.com
R329.21
A
10.3969/j.issn.1000- 4718.2016.09.013
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13 06:39:10
昆明医科大学学报(2021年1期)2021-02-07 01:06:50
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年8期)2017-11-22 03:18:21
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
兽医导刊(2016年6期)2016-05-17 03:50:50
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
长江蔬菜(2015年3期)2015-03-11 15:10:29
当代畜禽养殖业(2014年5期)2014-08-31 02:50:50
