
固相萃取—气相色谱—串联质谱法同时测定人体尿液中4种有机磷农药代谢物
2016-10-21 11:35黄梦莹王娜郭欣妍邱盼子汤卫国张尊建
分析化学 2016年5期
黄梦莹 王娜 郭欣妍 邱盼子 汤卫国 张尊建
摘要:建立了同时测定尿液中4种有机磷类农药代谢物的气相色谱/串联质谱分析方法。尿液样品以WCX固相萃取柱富集提取,乙酸乙酯乙腈(70∶30, V/V)再萃取,浓缩干燥,甲苯溶解,N叔丁基二甲基甲硅烷基N甲基三氟乙酰胺衍生化。采用HP5MS色谱柱(30 m×0.25 mm×0.25 μm)程序升温分离,串联质谱多反应检测,内标法定量。通过比较尿样中代谢物在不同萃取溶剂、固相萃取柱及pH值洗脱液等条件下的回收率,优化了前处理方法。在0.2~200 μg/L范围内,4种代谢物峰面积与内标峰面积的比值与质量浓度的线性关系良好(R2≥0.992),方法检出限为0.083~0.667 μg/L,定量限为0.2~2.0 μg/L,平均回收率为54.1%~68.6%,相对标准偏差均小于8.5% (n=6)。本方法稳定、可靠,分析时间短,不需要使用大量有机溶剂,可同时处理大批量样品,适用于人群有机磷类农药暴露情况的评估。
关键词 :固相萃取; 气相色谱串联质谱; 有机磷农药代谢物; 尿液
1 引 言
继有机氯农药被全面禁用后,有机磷类農药(Organophosphorus pesticides,OPs)成为国内目前农业生产上应用最广泛、使用量最大的农药[1]。OPs具有易降解、杀虫效果好、抗性不显著、残效期较短的特点,除广泛应用于农业生产中[2],还常作为除草剂应用于公园、绿地等公共场所。
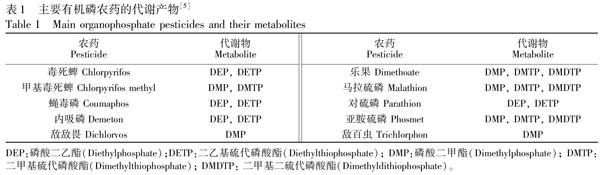
OPs进入人体后代谢迅速,蓄积现象不明显。OPs由于其结构的相似性,进入体内后一般都能代谢成为6种二烷基磷酸酯(DialkylPhosphates,DAP)代谢产物中的1种或几种,在较短的时间内随尿排出[3],主要有机磷农药代谢产物见表1。除6种二烷基磷酸酯外,OPs还会产生特殊代谢产物,每种特殊代谢产物通常只由特定的1种或几种OPs代谢产生,如毒死蜱、甲基毒死蜱的特殊代谢产物为3,5,6三氯2吡啶醇(3,5,6TCP),对硫磷、甲基对硫磷的特殊代谢产物为对硝基酚,马拉硫磷的特殊代谢产物为马拉硫磷二羟基酸[4]。
近年来,农药暴露对人体健康的影响受到广泛关注[6~9]。根据OPs代谢较快的特点,检测人群尿液样品中OPs非特异性及特异性代谢物含量可有效评估人体的OPs暴露水平。检测尿液中OPs代谢物的方法主要有气相色谱质谱联用法[10~12]、气相色谱串联质谱法[13~15]、液相色谱串联质谱法[16~18]。液相色谱串联质谱法前处理较为简单,但实际应用中灵敏度不高,且基质效应造成的离子抑制比较普遍[19]。提取尿液中DAPs的方法主要有液/液萃取[10~12,15~17]、固相萃取[13,20]、冻干法[14]。本研究采用固相萃取液/液萃取气相色谱串联质谱技术建立了人体尿液中3种OPs非特异性代谢产物及1种特殊代谢产物的高灵敏检测方法,避免了液/液萃取使用乙醚对实验室造成的安全隐患,实验时间大幅缩短,净化效率优于冻干法。本方法为有机磷农药的人体内暴露研究提供了良好的技术支撑。
2 实验部分
2.1 仪器与试剂
Quattra micro GCMS/MS(美国Waters公司);AG285电子天平(瑞士Mettler公司); MG2200氮吹仪(日本EYELA公司);R210旋转蒸发器(瑞士Buchi公司);WH3微型旋涡混合仪(上海楚定分析仪器有限公司,中国);Centrifuge C5804离心机、恒温混匀仪(德国 Eppendorf公司);MilliQ超纯水器(美国Mimpore公司)。
乙腈、乙酸乙酯、甲苯(分析纯,南京化学试剂有限公司);甲醇、甲酸(色谱纯,德国Merck公司);N叔丁基二甲基甲硅烷基N甲基三氟乙酰胺(MTBSTFA,>97.0%,美国Aldrich公司)。 其它试剂均为国产分析纯。
Waters Oasis WCX固相萃取柱(150 mg,6 mL)、Waters Oasis HLB固相萃取柱(200 mg,6 mL)、Waters Oasis WAX固相萃取柱(60 mg,3 mL),均购自美国Waters公司。
固相萃取DAPs:准确吸取尿液10.0 mL于100 mL具塞塑料离心管中,加入100 μL浓HCl,涡旋1 min,上样于Waters Oasis WCX固相萃取柱(柱子提前用6 mL甲醇、6 mL水活化),通过固相萃取小柱的尿液样品收集于100 mL塑料离心管中。上样后,干燥,用8 mL 10%甲酸甲醇溶液洗脱,洗脱液置于10 mL具塞比色管中。
液/液萃取TCP:通过固相萃取小柱的尿液样品中加入适量NaOH,使样品pH≈7.0,加入2 g NaCl,涡旋至溶解,加入20 mL乙酸乙酯乙腈(70∶30, V/V),涡旋3 min,6000 r/min离心5 min。上清液经过含适量无水Na2SO4的漏斗转移至鸡心瓶中,旋蒸浓缩至2 mL左右。
用移液器将萃取浓缩液移出至含洗脱液的比色管中,40℃水浴中氮吹至完全干燥,比色管中加入0.5 mL甲苯,涡旋1 min,转移至1.5 mL玻璃小管中,加入10 μL 0.2 mg/L内标DBP,20 μL MTBSTFA,置于90℃恒温混匀仪中衍生化30 min。
衍生化后的溶液冷却至室温,过0.22 μm有机相滤膜,供GCMS/MS分析。
2.3 气相色谱条件
DB5MS色谱柱(30 m×0.25 mm×0.25 μm);程序升温:60℃保持2 min,以5℃/min升至90℃,以12℃/min升至160℃,再以10℃/min升至280℃,保持2 min;进样量1.0 μL;不分流进样,载气:氦气(99.999%),流量:1.0 mL/min,进样器温度: 250℃。
2.4 质谱条件
正离子模式的电喷雾电离ESI+,多反应离子监测(MRM),TCP采用选择离子检测(SIM), EI源轰击能:70 eV,离子源温度:180℃,传输线温度:250℃,溶剂延迟时间:5.0 min,碰撞气:高纯氩气,保留时间定性,内标法定量。其它质谱参数见表2。
3 结果与讨论
3.1 空白基质的选择
选择空白基质时,如采用人工合成尿液进行实验,虽然本底较为干净,干扰小,但人工尿液中不含蛋白质,与实际尿液有很大差距,不能有效反映人体实际尿液基质的复杂性。如采用单个个体尿液作为空白基质,有的个体存在较高的本底值,不适合测定方法的建立。因此,本实验通过筛选不同个体的尿液,选择本底干扰较小的人体尿液作为本实验的空白基质。
3.2 前处理条件优化
3.2.1 液/液萃取 大量文献报道DAPs和TCP提取均采用液/液萃取的方法,故本实验首先采用液/液萃取的方法提取DAPs和TCP。萃取溶剂见表3,结果见图2。结果表明,采取液/液萃取法提取DAPs时,同一提取方案对不同代谢物提取回收率差别较大,很难找到平衡方案使各代谢物的回收率均符合要求,TCP的液/液萃取回收率較高。由于TCP的pKa=7.5,近中性,故萃取时将溶液调至pH ≈7,确保萃取时TCP的存在形式为分子态[18]。
3.2.2 固相萃取柱的选择
目前文献所报道的DAPs和TCP提取多采用液/液萃取法,固相萃取法的报道较少。本实验分别考察了亲水亲油平衡柱(Waters Oasis HLB)、弱阳离子交换柱(Waters Oasis WCX)及弱阴离子交换柱(Waters Oasis WAX)对各代谢物的萃取效果,结果见图3。
Waters Oasis HLB柱虽然适用范围较广,可从各种基质中分离出多种酸性、碱性和中性化合物,但DMTP回收率过高,其它物质则过低。Waters Oasis WAX柱为弱阴离子交换柱,适用于酸性物质, 具有高选择性,DAPs为酸性(DMP pKa=1.25, DMTP pKa=1.37, DEP pKa=1.37, DETP pKa=1.49),但本实验发现其对DMP、DEP基本没有保留,对DMTP、DETP虽有保留,但回收率非常不稳定,不适用于从尿液中提取DAPs,而TCP的pKa近中性,不易被WAX柱吸附。
Waters Oasis WCX柱对DMTP、DEP、DETP、TCP均有保留,且回收率较稳定。Waters Oasis WCX柱为阳离子交换柱,可吸附带正电的分子。DAPs的pKa较低,呈酸性,实验中采用强酸酸化并不会使DAPs分子解离,而分子中的硫原子及氧原子含有孤对电子,吸引溶液中的氢质子,从而使DAPs分子质子化,带正电荷,可被WCX柱吸附。DMP相比于其它的DAPs,由于极性较大,正电性较弱,在WCX柱上基本无保留。WCX柱对除DMP外的DAPs及TCP均有保留,故选其作为固相萃取柱,并对其洗脱条件进行优化。
3.2.3 洗脱条件的优化 在洗脱条件的优化中,分别考察10%甲酸甲醇溶液、20%甲酸甲醇溶液、30%甲酸甲醇溶液,10%甲酸乙腈溶液的洗脱效果,结果见图4。使用10%甲酸甲醇作为洗脱液时,各代谢物回收率在34.9%~77.5%之间(DMP基本无回收),高于20%甲酸甲醇溶液得到的回收率,且较30%甲酸甲醇溶液得到的回收率更为稳定。10%甲酸乙腈溶液的洗脱效果与10%甲酸甲醇溶甲醇溶液相当,TCP基本无回收。随着酸度增加,DAPs的回收率基本都是呈先降低再升高的趋势。使用10%甲酸甲醇作为洗脱液,回收率可以接受,且较为稳定。
3.3 固相萃取与液/液萃取的比较
采用表3中方案4作为液/液萃取方法,与优化过的固相萃取方法进行回收率的比较,结果见图5。
对于DMTP, DEP和DETP,固相萃取的回收率和稳定性均高于液/液萃取。对于生物基质样品分析,方法的稳定性十分重要。液/液萃取提取TCP的回收率和稳定性优于固相萃取。用何种提取方法DMP,结果均不能令人满意。最终采用固相萃取提取DMTP, DEP和DETP,采用表3中方案4提取TCP。
3.4 衍生化反应
本实验采用硅烷化试剂N(叔丁基二甲基硅烷基)N甲基三氟乙酰胺(MTBSTFA)对目标化合物进行衍生化反应。目标代谢物与衍生化试剂MTBSTFA的反应原理如图6所示,通过硅烷化作用将叔丁基二甲基硅烷基引入目标代谢物分子中,取代分子中的活性氢。
文献[5,10,11,20,21]采用五氟溴苄苯(PFBBr)对DAPs进行衍生化,衍生化反应需加入适量K2CO3。与采用PFBBr相比,MTBSTFA的衍生化反应时间较短(PFBBr衍生化反应需16 h[21]),不需要其它试剂参与,不必考虑其它试剂的用量对衍生化效率的影响。
3.5 方法学验证
3.5.1 标准曲线及线性范围 向空白尿样中添加目标代谢物混合标准溶液,按2.2节中前处理方法进行提取、净化,在优化后的色谱及质谱条件下进样,绘制工作曲线,记录各待测物色谱峰面积(As)与内标色谱峰面积(Ar)。以各待测物与内标的峰面积比R (R= As/As)对浓度(C,mg/L)进行线性回归,确定线性范围及检出限、定量限。检出限和定量限分别以3和10倍信噪比的浓度来确定。DMP在固相萃取柱上基本无保留,其它代谢物制定工作曲线得到的线性方程相关系数均在0.9923~0.9942之间,表明各代谢物在相应的浓度范围内线性关系良好, 结果见表4。
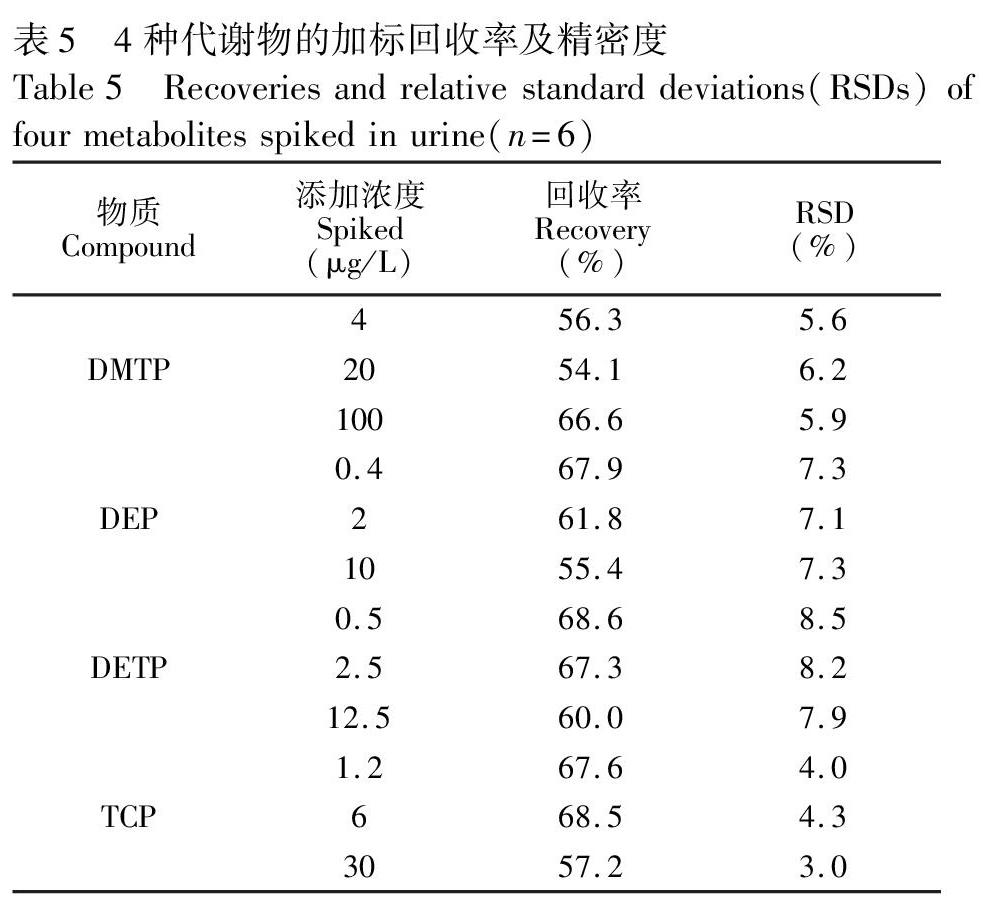
3.5.2 方法的回收率及精密度 由于无法获得所分析的代谢物引入叔丁基二甲基硅烷基的标准品,衍生化反应效率无法确定,整个样品制备过程的绝对回收率无法考察。本研究按2.2节方法同时处理加标与未加标的空白基质,未加标空白基质在衍生化前加入等量标准溶液,由两者响应的比值计算回收率,设置低、中、高3个添加浓度,结果见表5。生物体液样本基质较为复杂,分析前需要经过衍生化反应,处理步骤较多,回收率低于基质单一的样品。
3.5.3 基質效应 采用提取后添加法评定基质效应。按照2.2节方法处理空白尿液基质,衍生化前加入标准品溶液,进样后所得峰面积记为A组峰面积;纯溶剂衍生化前加入标准品溶液,进样后所得峰面积记为B组峰面积。以A组峰面积与B组峰面积的比值(A/B)考察绝对基质效应,4种代谢物的绝对基质效应在60%~176%之间,故采用基质加标工作曲线消除基质效应的影响。
3.6 气相色谱质谱条件优化
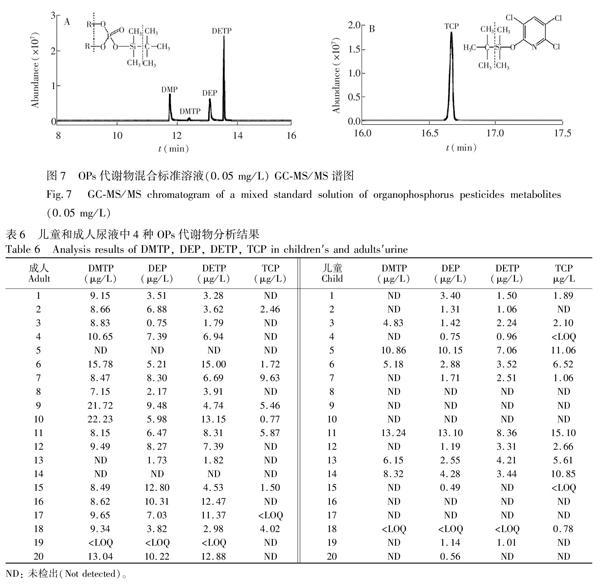
根据目前已有文献报道,本研究尝试了不同的升温程序。优化后的色谱图见图7,各物质峰形良好,分离效果理想。
代谢物衍生化后在EI源中主要的裂解方式见图7,运用Daughter Scan模式,进行子离子扫描,并对碰撞能量进行优化,以达到较高的灵敏度。3,5,6TCP在正离子检测方式下,基峰为m/z 256,运用Daughter Scan模式,进行子离子扫描, 结果并未发现响应稳定且较强的碎片峰,这与3,5,6TCP的环状结构特点较为一致。
3.7 样品分析
采集居住在农药厂附近居民的晨尿样品40份,其中儿童尿液样品20份,成人尿液样品20份,按2.2节中的实验方法进行处理,测定结果见表6。
结果表明,成人尿液样品DMTP, DEP, DETP, TCP检出率分别为85%, 90%, 90%, 45%,儿童尿液样品分别为35%, 75%, 65%, 60%。成人尿液中非特异性代谢物DMTP, DEP, DETP的检出率均高于儿童,特异性代谢物TCP的检出率低于儿童。结果表明,居住在农药厂附近的成人与儿童均有不同程度的有机磷农药内暴露,呼吸可能是造成有机磷农药内暴露的重要途径。以尿液中代谢物作为评估有机磷农药暴露程度的指标,成人的暴露量大于儿童,这是由于成人的呼吸量较大, 且对农药的代谢能力较强。不同成人与儿童尿液中有机磷代谢物检出的差异与其住所位置、膳食结构等都有密切的关系。
4 结 论
建立了固相萃取气相色谱串联质谱同时分析制尿液中4种OPs代谢物的方法,定量检测了人体尿液中4种代谢物含量。本研究采用WCX固相萃取柱,与液/液萃取相比,耗费的有机溶剂更少,耗时更短,适合于大批量样品的前处理,为研究人群中低剂量OPs农药暴露情况提供了方法学基础。
Reference
1 ZHU XiaoLan, CAI JiBao, YANG Jun, SU QingDe. Chinese J. Anal. Chem., 2005, 33(6): 821-824
朱晓兰, 蔡继宝, 杨 俊, 苏庆德. 分析化学, 2005, 33(6): 821-824
2 WU Yan, KANG QingHe, GAO KaiYang, LI ZhiBin. Chinese J. Anal. Chem., 2009, 37(5): 753-757
吴 岩, 康庆贺, 高凯扬, 李志斌. 分析化学, 2009, 37(5): 753-757
3 Koureas M, Tsakalof A, Tsatsakis A. Hadjichristodoulou. Toxicol Lett., 2012, 210: 155-168
4 LIU Yong, TANG YingFei, SONG JinFeng, HU ZhiWei. Chinese Journal of Chromatography, 2014, 32(2): 139-144
刘 永, 唐英斐, 宋金凤, 胡志伟. 色谱, 2014, 32(2): 139-144
5 YANG YuLin, RUI ZhengRong, WANG Hong, WANG GuoQuan, CHEN Jia, ZHOU ZhiJun. Chinese J. Health Lab. Technol., 2003, 13(1): 24-26
杨玉林, 芮振荣, 王 宏, 汪国权, 陈 嘉, 周志俊. 中国卫生检验杂志, 2003, 13(1): 24-26
6 Engel S M, Wetmur J, Chen J, Zhu C B, Barr D B, Canfield R L, Wolff M S. Environ. Health Perspect., 2011, 119(8): 1182-1188
7 CHEN Chao, CHEN GuoQing, GAO ShuMei, KONG FanBiao, LI Run, HUANG QiFeng. Spectroscopy and Spectral Analysis, 2012, 32(6): 718-722
陈 超, 陈国庆, 高淑梅, 孔凡标, 李 润, 黄奇峰. 光谱学与光谱分析, 2012, 32(6): 718-722
8 Rauh V, Arunajadai S, Horton M, Perera F, Hoepner L, Barr D B, Whyatt R. Environ. Health Perspect., 2011, 119(8): 1196-1201
9 Ding G D, Shi R, Gao Y, Zhang Y, Kamijima M, Sakai K, Wang G Q, Feng C, Tian Y. Environ. Sci. Technol., 2012, 46: 13480-13487
10 Prapamontol T, Sutan K, Laoyang S, Hongsibsong S, Lee G, Yano Y, Hunter R E, Ryan P B, Barr D B, Panuwet P. Int. J. Hyg. Environ. Health, 2014, 217(45): 554-566
11 Ueyama J, Kamijima M, Kondo T, Takagi K, Shibata E, Hasegawa T, Wakusawa S, Taki T, Gotoh M, Saito I. J. Chromatogr. B, 2010, 878: 1257-1263
12 Ueyama J, Saito I, Kamijima M, Nakajima T, Gotoh M, Suzuki T, Shibata E, Kondo T, Takagi K, Miyamoto K, Takamatsu J, Hasegawa T, Takagi K. J. Chromatogr. B, 2006, 832: 58-66
13 Alwis G K H D, Needham L L, Barr D B. J. Chromatogr. B, 2006, 843: 34-41
14 Bravo R, Caltabiano L M, Weerasekera G, Whitehead R D, Fernandez C, Needham L L, Bradman A, Barr D B. J. Expo. Anal. Environ. Epidemiol., 2004, 14: 249-259
15 Hill Jr. R H, Shealy D B, Head S L, Williams C C, Bailey S L, Gregg M, Baker S E, Needham L L. J. Anal. Toxicol., 1995, 19: 323-329
16 Dulaurent S, SaintMarcoux F, Marquet P, Lachatre G. J. Chromatogr. B, 2006, 831: 223-229
17 Bicker W, Lammerhofer M, Lindner W. J. Chromatogr. B, 2005, 822: 160-169
18 WANG Na, SUN Juan, SHI LiLi, JI GuiXiang, CHEN GuoSong. Chinese Journal of Chromatography, 2013, 31(9): 903-907
王 娜, 孫 娟, 石利利, 吉贵祥, 陈国松. 色谱, 2013, 31(9): 903-907
19 Odetokun M S, Montesano M A, Weerasekera G, Whitehead Jr. R D, Needham L L, Barr D B. J. Chromatogr. B, 2010, 878: 2567-2574
20 Alwis G K H D, Needham L L, Barr D B. Talanta, 2009, 77: 1063-1067
21 WU ChunHua, ZHENG LiXing, ZHOU ZhiJun. Fudan Univ. J. Med. Sci., 2006, 33(4): 552-555
邬春华, 郑力行, 周志俊. 复旦学报医学版, 2006, 33(4): 552-555
Abstract A method for quantifying four urinary metabolites of organophosphorus pesticides using gas chromatographytandem mass spectrometry (GCMS/MS) was developed. The metabolites were extracted and enriched from urine samples with WCX solid phase extraction (SPE) cartridges, followed by liquidliquid extraction with ethyl acetateacetonitrile (70∶30, V/V). The analytes were chemically derivatized with NtertbutyldimethylsilylNmethyltrifluoroacetamide after the derivatives were concentrated by drying and dissolved in phenylmethane. The separation was performed on a HP5MS capillary column (30 m×0.25 mm×0.25 μm) with temperature programming and the detection was performed in multiple reaction monitoring (MRM) mode using GCMS/MS. The internal standard method was used for quantification. The extraction solvents, types of SPE cartridges and eluents were optimized by comparing the sample recoveries under different conditions. The result showed that the calibration curves of four metabolites were linear in the range of 0.2-200 μg/L (R2≥0.992). The limits of detection (LODs) and the limits of quantification (LOQs) of the four metabolites were 0.083-0.667 μg/L and 0.2-2.0 μg/L, respectively. The recoveries of four metabolites ranged from 54.1% to 68.6% (RSD<8.5%, n=6). The established method whichs avoid using large amount of organic solvents in liquidliquid extraction is stable, reliable and efficient, and is suitable for the analysis of large amounts of samples. Therefore, the method can be applied to assess the exposure level of organophosphorus pesticide in general population.
Keywords Solid phase extraction; Gas chromatographytandem mass spectrometry; Organophosphorus pesticides metabolites; Urine
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
医学概论(2021年9期)2021-09-16
养生保健指南(2019年8期)2019-12-18
时代英语·高一(2018年4期)2018-09-14
当代工人·精品C(2016年6期)2017-01-12
分析化学(2016年7期)2016-12-08
肉类研究(2015年5期)2015-08-08
肉类研究(2015年1期)2015-04-08
为了孩子(孕0~3岁)(2000年3期)2000-06-13
