
液相色谱-串联质谱法测定豆制品中的乌洛托品
2016-09-15 16:04李新丽张厚森朱成晶
山东化工 2016年14期
李新丽,张厚森,贾 涛,高 宏,朱成晶
(江苏省理化测试中心,江苏 南京 210042)
液相色谱-串联质谱法测定豆制品中的乌洛托品
李新丽,张厚森,贾 涛,高 宏,朱成晶
(江苏省理化测试中心,江苏 南京 210042)
建立了用液相色谱-串联质谱法测定豆制品中乌洛托品含量的方法。样品用乙腈提取、正己烷净化,弱碱性流动相、梯度洗脱、正离子电喷雾多反应监测(MRM)模式检测。本方法适用于腐竹、大豆粉、豆浆粉、大豆蛋白粉等豆制品中乌洛托品的测定。方法定量限为4 μg/kg,回收率86.2 %~102.0 %,精密度RSD<5 %。实验表明本方法前处理简单,定性定量方法准确,能对豆制品中乌洛托品进行确证。
乌洛托品;液相色谱串联质谱法;测定
乌洛托品(别名六亚甲基四胺)是卫生部公布的非食用物质之一,在弱酸条件下分解产生甲醛,具有杀菌、防腐的作用。卫生部报道可能有些不法厂家违法使用乌洛托品以替代吊白块,意在保持产品的色泽和延长保质期。乌洛托品本身属低毒类,可作为药物服用,但其在酸性条件下,分解出的甲醛易与体内多种化学结构的受体发生反应,如与氨基化合物可以发生缩合,与巯基化合物加成,使蛋白质变性。甲醛在体内还可还原为醇,故可表现出甲醇的毒理作用。对人体的肾、肝、中枢神经、免疫功能、消化系统等均有损害。因为乌洛托品在pH小于6.5就会分解放出甲醛,在食品中的残留量会随着存放时间的延长而减少,在市场调查收集的阳性样品中乌洛托品的残留量都在10 mg/kg以下。卫生部发布的第四批《食品中可能违法添加的非食用物质和易滥用的食品添加剂品种名单》中明确规定乌洛托品属于违法添加的非食用物质。目前现有食品中乌洛托品测试的国标方法有SN/T 2226-2008《进出口动物源性食品中
乌洛托品残留量的检测方法》[1],适用于鸡肉、鸡肝脏、鸡肾脏和猪肉中乌洛托品残留量的检测和确证,因为样品基质差别太大,该检测方法不适用豆制品。报道的食品中乌洛托品的检测主要有气相色谱法[2-3]、分光光度法[4-5]、离子色谱法[6]、液质法[7-17]等,本文介绍的方法前处理更为简单、快速。和同类液质法相比,因为采用梯度洗脱分离了豆制品中的干扰物质,从而简化了目标物提取过程中去杂质的步骤,降低了检测成本、提高了检测速度。
本文通过对不同提取方法、提取溶剂、色谱条件的研究,建立了用乙腈震荡提取、正己烷净化,弱碱性流动相、梯度洗脱的液相色谱串联质谱法测定豆制品中乌洛托品残留量的分析方法,该方法操作过程简单,准确度、灵敏度和精密度好。
1 实验部分
1.1 试剂与材料
乌洛托品(纯度99.0 %) 德国Dr. Ehrenstorfer公司;氨水,正己烷,甲酸铵均为分析纯,国药集团;乙腈为色谱纯, 美国TEDIA 公司; 0.22 um微孔滤膜,天津津腾实验设备有限公司。
1.2 仪器与设备
高压液相色谱三重四级杆串联质谱仪(HPLC-MS/MS),型号Agilent1260-6430 美国安捷伦公司;高速离心机;电动振荡器;涡旋混匀器。
1.3 液相色谱与质谱条件
1.3.1 色谱柱为科奥美萃生物科技公司生产的Kinetex HILIC 100A 柱(50 mm*2.1 mm,2.6 um);流动相为10 mmol/L甲酸铵溶液(pH=7.5±0.3),乙腈;色谱柱温度25℃;进样量5μL。梯度洗脱程序见表1。
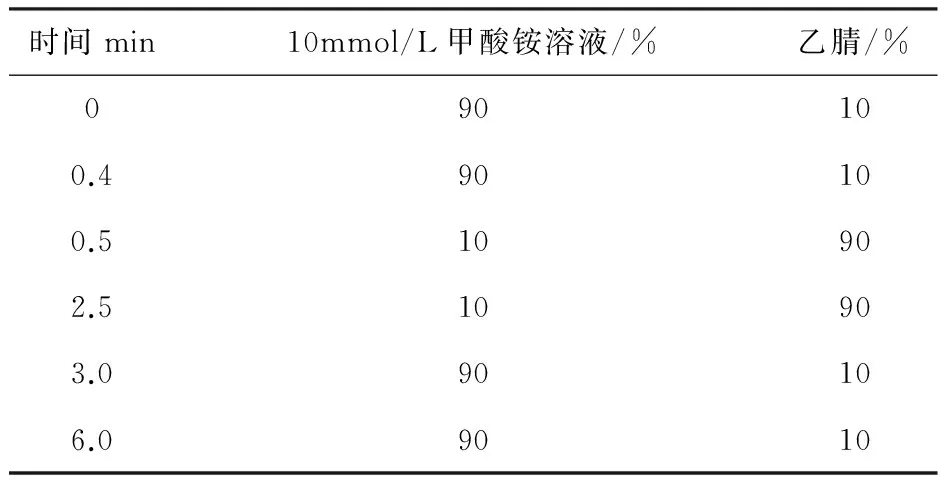
表1 流动相及梯度洗脱条件
1.3.2 质谱条件
质谱扫描采集方法为多反应监测(MRM);质谱离子源为电喷雾(ESI)电离源,正离子模式,脱溶剂气为氮气,温度300 ℃,流量9 L/min;喷雾器压力103.4KPa;碰撞气为高纯氮气;毛细管电压4000 V;其它质谱条件见表2。

表2 乌洛托品多反应监测条件
1.4 试验方法
1.4.1 试样预处理
取有代表性的样品约200 g,用粉碎机粉碎,混匀,均分成两份作为试样,分别装入洁净容器,密封并做好标识,于-18 ℃冰箱内保存。
1.4.2 提取
准确称取试样2 g(精确至0.001 g),于50 mL具塞离心管中,用移液管准确加入10 mL乙腈,涡旋混匀,于震荡器(震荡频率300次/分)上震荡提取20min,以4000 r/min离心2 min,吸取上清液2 mL于10 mL离心管中,加入2 mL乙腈饱和的正己烷溶液,涡旋混匀1 min ,在4000 r/min下离心1 min,弃去上层正己烷,重复用乙腈饱和的正己烷溶液萃取一次,下层液过0.22 μm滤膜,待测。
1.5 标准溶液配制
准确称取乌洛托品标准品,用乙腈配制成浓度为100 μg/mL的标准储备液。储备液在0 ℃~4 ℃冰箱避光保存,可使用6个月。使用前用乙腈配制浓度为0.0 ng/mL、5.0 ng/mL、10.0 ng/mL、40 ng/mL、80 ng/mL、100 ng/mL的标准工作液。
1.6 测定
1.6.1 定性测定
按照上述条件测定样品和标准溶液,如果样品与标准溶液中待测物的质量色谱峰保留时间一致;且在扣除背景后的样品质量色谱图中,所选择的离子对均出现,同时与标准品的相对丰度允许偏差不超过表3规定的范围,则可判定为样品中存在对应的待测物。

表3 定性确证时相对离子丰度的最大允许偏差
1.6.2 定量测定
以标准使用液分别进样,以峰面积为纵坐标,标准溶液浓度为横坐标绘制标准工作曲线,用标准工作曲线对样品进行定量,样液中乌洛托品的响应值应在标准使用液线性范围内。
2 结果与讨论
2.1 前处理方法选择
在选择提取溶剂时考察了水、甲醇、乙醇、乙腈等溶剂,水的提取液浑浊,很难分离出澄清液,容易发生堵塞;乙醇能和正己烷互溶,影响脂溶性杂质的分离,增加基质效应;甲醇和乙腈的提取效率相近,考虑甲醇的沸点低和毒性较大,故选择乙腈。
考察了超声提取和震荡提取法,发现超声提取会使目标物发生分解,回收率降低、且数据不稳定;考察了震荡提取的震荡频率、提取时间和提取次数等因素,用阳性样品实验发现,当震荡频率300 次/min时,提取20 分钟一次就能达到最大提取值,加标回收率90 %以上、精密度也能满足要求。
在净化方法的选择上,考察了固相萃取小柱净化和正己烷净化,参照SN/T2226-2008,采用离子交换树脂小柱,但因为豆制品中可溶于乙腈的基质复杂,采用小柱并不能消除基质干扰,且因为每个小柱的填充差异,影响了方法的精密度和准确性,选择正己烷净化不但步骤简单,成本低,而且消除基质效应比小柱好。
2.2 仪器条件选择
乌洛托品在酸性条件下会分解放出甲醛,采用碱性甲酸铵缓冲盐溶液和乙腈作为流动相不仅可以为质谱的电离源提供足够的带电离子而且可以将整个分离过程维持在一个相对稳定的碱性环境中,可以避免乌洛托品在酸性条件下的分解。
实验表明,采用电喷雾正模式电离、碱性色谱条件下比酸性条件下的色谱峰响应值高。
豆制品特别是腐竹经过了复杂加工后,基质复杂,对乌洛托品检测干扰很大,即使在前处理中用正己烷进行净化,但基质效应仍然较大,而且相同种类不同品牌豆制品的基质效应也差别较大,为此考察了不同比例流动相的等度洗脱、梯度洗脱、不同流速的选择等。实验表明当进样量5 μL、流速0.2 mL/min、梯度洗脱时可以消除所有基质干扰,用乙腈配置标准曲线,基质提取液加标回收率达到95 %~100 %。

图1 样品中乌洛托品的总离子流图

图2 乌洛托品标准品的总离子流图

图3 乌洛托品质谱图
2.3 方法的线性关系和检出限
对系列标准溶液进行测定,在5 μg/kg ~100 μg/kg范围内线性良好,相关系数r2大于0.999。
在空白样品基质溶液中加入估计检出限浓度(大于3倍信噪比)的标准品进行检出限测试,结合回收率确定腐竹、大豆粉、豆浆粉、大豆蛋白粉等豆制品中乌洛托品的检出限小于1 μg/kg。
2.4 方法回收率和精密度
在阴性样品中加入乌洛托品标准物质,添加的标准品使测试溶液浓度分别为5 μg/L、10 μg/L、50 μg/L,进行回收率和精密度实验。腐竹因为基质复杂,不同品牌基质差异较大,实验过程选择3个不同产地和品牌的腐竹样品进行试验。试验表明方法精密度RSD=0.69%~2.89%,方法回收率为88.8%~102.4%。实验结果见表4。

表4 精密度和准确度测试数据(平行测定次数n=6)
3 结论
对市售腐竹阳性样品研究发现,在常温下乌洛托品在缓慢分解,在-18℃下储存可停止分解,从获得的阳性样品的检测数据看,乌洛托品的残留量都在1 mg/kg以上,本法能满足豆制品中违法添加乌洛托品的有效检出,可以在豆制品的食品安全监管以及监督中发挥作用。
[1] 中华人民共和国国家质量监督检验检疫总局,SN/T 2226-2008进出口动物源性食品中乌洛托品残留量的检测方法液.
[2] 黄国春.气相色谱法测定腐竹中乌洛托品含量的研究[J].广西轻工业,2008,6(6):26.27.
[3] 王庭欣,等,腐竹中乌洛托品的高效气相色谱检测技术[J].河北大学学报:自然科学版,2013,33(5):477-483.
[4] 魏运金,粮食制品中雕(吊)白块六亚甲基甲四胺的检测[J].福建轻纺,2004,183(8):16-19,33.
[5] 唐红霞,等.分光光度法检测腐竹中乌洛托品[J].粮油食品科技,2014,22(5);62-65.
[6] 张社利,等.离子色谱法检测腐竹、粉丝中的乌洛托品[J].应用化学,2014,31(11);1352-1355.
[7] 温 韬,等.高效液相色谱法测定腐竹中的乌洛托品[J].洛阳理工学院学报:自然科学版, 2013,23(3);1-4.
[8] 周秀云,等.SPE-UPLC-MS/MS快速测定豆制品中的乌洛托品[J].河北省科学院学报,2013,30(1);64-68.
[9] 汪 辉,等.液相色谱-串联质谱法快速测定食品中的乌洛托品[J].食品与机械, 2013,29(3);72-78.
[10] 乔 玲,等.凝胶净化色谱-快速液相串联质谱法测定豆制品中乌洛托品的含量[J].食品科技,2014,39(5);275-278.
[11] 杨 芳,等.高效液相色谱-串联质谱法测定食品中的乌洛托品[J].食品工业,2014,35(9);278-281.
[12] 张爱芝,张书芬,王全林,沈 坚.超高压液相色谱-串联质谱法测定腐竹、米线、年糕中的乌洛托品[J].食品安全质量检测学报,2013,4(2):472-478.
[13] 冼燕萍,l等.UPLC-MS/MS测定腐竹和米粉中的乌洛托品[J].江南大学学报,20l2, 11(1),78-82.
[14] 高 洁,等.离子交换固相萃取-超高效液相色谱-串联质谱法快速测定腐竹中的乌洛托品[J].食品安全质量检测学报, 2014,5(10):3210-3214.
[15] 张厚森,等.高效液相色谱-串联质谱法测定腐竹中乌洛托品残留量[M].理化检验:化学分册,2014,50(10):1245-1248.
[16] 马雪涛,等.离子交换固相萃取结合超高效液相色谱-串联质谱法测定豆制品中乌洛托品残留量[J].食品科学,2014,35(10);166-169.
[17] 徐 幸,等.高效液相色谱-稳定性同位素稀释质谱法测定米线中乌洛托品的不确定度评[J].食品科学,2015, 36(16):246-250.
(本文文献格式:李新丽,张厚森,贾 涛,等.液相色谱-串联质谱法测定豆制品中的乌洛托品[J].山东化工,2016,45(14):63-66.)
Determination of Urotropine content in Bean Products by HPLC-MS/MS
LiXinli,ZhangHousen,JiaTao,GaoHong,ZhuChengjing
(Physics and Chemistry Testing Center of Jiangsu Province,Nanjing 210042, China)
Determination content of Urotropine in Bean Products is descripted in this paper. The samples were extracted by acetonitrile, purified by hexane, gradient elution separated by weak basic mobile phase, and determined by electrospray ionization and positive ion monitoring mode. This method can used to determine the content of Urotropine in beancurd skin, soybean flour, soymilk powder, and soyabean protein powder. The limitation is 4μg/kg, the total recovery rate is in the range of 86.2 %~102.0 %, and RSD<5 %. Its advantages include simple pretreatment and high accuracy.
Urotropine, HPLC-MS/MS ,Determination
2016-05-11
食品安全地方标准JSSPDB2012-004
李新丽(1968—),女,高工,从事分析测试与研究工作。
O657.63;TS207.3
A
1008-021X(2016)14-0063-04
猜你喜欢
江苏卫生保健(2021年12期)2022-01-17
食品安全导刊(2021年21期)2021-08-30
保健与生活(2021年15期)2021-08-16
理化检验-化学分册(2020年12期)2021-01-26
农家之友(2019年3期)2019-01-16
中国生殖健康(2019年5期)2019-01-06
现代营销(创富信息版)(2018年7期)2018-09-05
现代营销(创富信息版)(2018年6期)2018-09-05
食品界(2018年8期)2018-09-03
现代营销(创富信息版)(2018年5期)2018-07-12
