
应用16S rRNA高通量测序法比较人与常用实验动物口腔菌群的异同
2016-09-13 05:04:55顾东曙陈傍柱刘海月那顺巴雅尔周宏伟顾为望
中国比较医学杂志 2016年8期
顾东曙,陈傍柱,江 霞,刘海月,那顺巴雅尔,周宏伟,顾为望
(1.南方医科大学实验动物中心,广州 510515;2.南方医科大学公共卫生与热带医学学院,广州 510515;3.广东医科大学,东莞 523808; 4.南方医科大学珠江医院检验医学部,广州 510282)
应用16S rRNA高通量测序法比较人与常用实验动物口腔菌群的异同
顾东曙1,陈傍柱1,江霞1,刘海月2,那顺巴雅尔3,周宏伟4,顾为望
(1.南方医科大学实验动物中心,广州510515;2.南方医科大学公共卫生与热带医学学院,广州510515;3.广东医科大学,东莞523808; 4.南方医科大学珠江医院检验医学部,广州510282)
目的应用16S rRNA高通量测序法测定几种常用实验动物(西藏小型猪、比格犬、猕猴、新西兰兔、Wistar大鼠)的口腔菌群,并和人的口腔菌群进行对比分析,为口腔微生态的动物模型研究提供基础性资料。方法用一次性棉拭子采集西藏小型猪、比格犬、猕猴、新西兰兔、Wistar大鼠和人的口腔菌群标本,提取样品总DNA,使用带标签的通用引物扩增16S rRNA V4区片段,Illumina 测序,经BIPES 以及QIIME 分析比较菌群多样性及结构。结果人与5种常用实验动物口腔菌群的丰富度均差异显著(P<0.05),不同种类动物有其各自独特的口腔菌群,但猴的口腔菌群与人最为相似。结论根据与人口腔某类菌群的相似程度,5种动物中,猴口腔的梭菌属(Fusobacterium)、卟啉菌属(Porphyromonas)水平与人最相似,提示猴可能是研究人口腔菌群较适宜的模型动物。从特定菌门角度,西藏小型猪可能是研究与变形菌门(Proteobacteria)相关疾病的较适宜模型动物;比格犬可能是研究与螺旋体门(Spirochaetes)相关疾病的较适宜模型动物。
实验动物;口腔菌群;16S rRNA;西藏小型猪;比格犬;猕猴;新西兰兔;Wistar大鼠
口腔菌群是由大量口腔微生物组成的复杂生态体系,口腔内适宜的温度、湿度、丰富的营养来源以及复杂的结构,为口腔内各种微生物的生长、繁殖和定居提供了便利条件,形成了口腔微生物的多样性[1]。口腔内微生物的多样性有利于维持口腔健康,抵御外界不良因素对机体的侵袭。一旦微生物群落失调,则可导致龋病、牙周病以及口腔溃疡等口腔疾病[2]。微生物的多样性及结构研究是进行复杂微生态系分析的基础,通过分析口腔中微生物丰度与结构的变化,能够揭示口腔疾病发生与微生物群落之间的关系,从而为预防和治疗口腔疾病提供合理的理论依据[3, 4]。细菌16S rRNA基因在所有细菌的基因组中存在,是编码rRNA相对应的DNA序列,在长时间的进化过程中,rRNA分子有些部位的序列变化非常缓慢,具有高度的保守性,基于这些序列的高度保守性可以检测细菌种系上的深远关系;同时,16S rRNA又具有9个高度可变区,V1至V9可变区(本研究中选择了V4可变区),可变区序列因细菌不同而异,保守区和可变区交错排列。通常基于保守区序列设计引物,PCR扩增,得到16S rRNA扩增片段,再利用可变区序列的差异实现对细菌种属的分类、鉴定。运用高通量测序技术测序,将高通量测序序列与基因数据库(或特定序列)进行比对,将一定相似度的菌属归为某种特定菌属,可实现对菌属快速、准确地分类鉴定,目前这种方法是研究口腔微生物多样性最为广泛,最为有效的手段之一。本研究通过课题组前期基于16S rRNA建立的Illumina 双末端测序法(BIPES)[4, 5],并采用QIIME比较分析几种常用实验动物与人的口腔菌群结构,为口腔疾病模型动物的选择提供数据基础。
1 材料和方法
1.1耗材
一次性棉拭子(苏州康健医疗器械有限公司)、各种型号EP管等耗材(上海Sangon公司)、口腔拭子基因组DNA快速提取试剂盒(北京百泰克生物技术有限公司)、Ex-taq酶(宝生物工程大连有限公司)。
1.2实验动物
成年普通级西藏小型猪(简称“猪”):东莞松山湖明珠实验动物科技有限公司提供(许可证号:SCXK(粤)2012-030);成年普通级猕猴(简称“猴”):南方医科大学实验动物中心提供(许可证号:SCXK(粤)2014-0025);成年普通级比格犬(简称“犬”):西安市迪乐普生物资源开发有限公司提供(许可证号:SCXK(陕)2014-001);成年SPF级新西兰大白兔(简称“兔”):南方医科大学实验动物中心提供(SCXK(粤)2011-0015);成年SPF级Wistar大鼠(简称大鼠):南方医科大学实验动物中心提供(SCXK(粤)2011-0015)。
1.3方法
1.3.1样品采集:一次性棉拭子采集成年猪、犬、猴、兔、大鼠口腔菌群样品,每个品种4只,雄性1只,雌性3只;40~60岁健康人群口腔样本10个,男性5个,女性5个。所有样品-20℃保存。
1.3.2样品总DNA提取:按照北京百泰克生物技术有限公司口腔拭子基因组DNA快速提取试剂盒说明书提取样品总DNA,置于-20℃保存。
1.3.3细菌16S rRNA基因扩增:选择带标签的通用引物扩增16S rRNA V4区域,引物由上海生工生物工程技术服务有限公司合成,引物上游:V4F-5′-GAGTGCCAGCMGCCGCGGTAA-3′,引物下游:V4R252-5′-TTAGGAGACCCGGACTACHVGGGTWT CTAAT-3′。每个样品对应相应的带标签上下游引物信息,PCR扩增体系为25 μL(表1)。PCR扩增条件:94℃,2 min;94℃,30 s;52℃,45 s;72℃,30 s;30个循环;72℃,5 min;4℃保存。
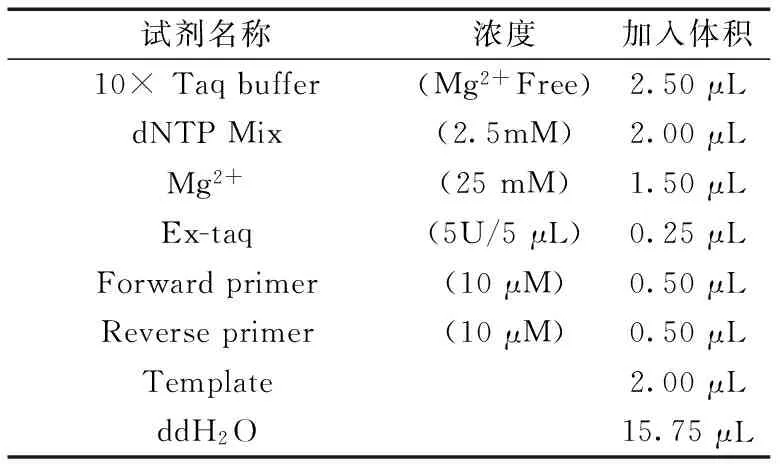
表1 PCR反应体系
1.3.4Illumina Solexa测序:为尽量避免因PCR扩增造成各样品浓度差异对后续基因测序深度的影响,样品上机测序之前进行凝胶电泳检测,利用凝胶分析软件BandScan以DNA Marker (2 000 bp) 200 bp条带为基准对DNA样品进行相对定量,确保尽量以相等的终溶度混合样品,然后将标准品(细菌质粒)加入样品PCR产物,以同等比例的终浓度混合均匀。Illumina HiSeq 2000测序平台进行高通量测序[5]。

图1 常用实验动物与人口腔菌群PD _whole_tree(A)和Shannon 指数(B)Fig.1 PD_whole_tree (A) and Shannon index (B) of oral flora in commonly used laboratory animals and humans
1.3.5数据分析:高通量测序数据用BIPES (Barcoded Illumina Paried-end Sequencing) 流程进行初步原始序列处理[6],。BIPES流程将碱基拼接和序列分析分为了两部分,一部分是使用mothur进行序列的挑拣,去除复杂的嵌合体序列,得到干净的序列,进而进入下一步的QIIME分析,包括alpha多样性,beta多样性和RDP聚类分析等。
使用TSC聚类将微生物进行界门纲目科属种的分类。并采用分阶段聚类算法RDP (Ribosomal Database Project, RDP)[7]将序列聚类成最小操作分类单元(Operational Taxonomic Units,OTUs),当细菌16S rRNA基因序列之间的距离在0.03以内,把它们视为同一个种属。对于获得的OTUs,使用对其进行分类注释。用mothur软件[8]进行测序数据的质量分析,去除错误序列和嵌合体。采用QIIME对样本PD_whole_tree、Shannon等多样性指数分析,从而分析样品的α多样性,即单个样品包含种属结构的多样性;基于UniFrac距离[8],进行主成分分析[9],分析β多样性;采用线性判别式分析效应值(linear discriminant analysis effect size,LEfSe)[10],Galaxy89在线工具寻找不同组别之间有统计学差异的菌群标志物。应用SPSS 20.0进行统计分析,采用One-Way ANOVA进行Bonferroni法两两比较分析或Kruskal-Wallis One-way ANOVA进行两两比较,校正检验水准。采用GraphPad prism5软件工具对PD_whole_tree、Shannon等作图。
2 结果
2.1测序与质量控制
35个样品质控后共获取了939272条16S rRNA基因序列,所有样品全部纳入后续分析。
2.2常用实验动物与人口腔菌群比较分析
2.2.1α多样性分析:α多样性通常是指群落内的物种丰度和均匀度。PD_whole_tree指观察到的种属数,反映菌落内的丰度;Shannon指数则受到菌落丰度和均度两者的影响。
图1 A显示:人与猪、犬、猴、兔、大鼠口腔菌群的α多样性,即菌群丰度差异显著(P<0.05);猪与犬、猴、兔口腔菌群的丰度差异显著(P<0.05),其他动物间均无显著差异(P>0.05);该结果提示,猪的口腔菌群多样性最低,人次之,其他4种动物口腔菌群物种丰富程度相对较高。图1 B显示:人、猪、犬、猴、兔、大鼠口腔菌群Shannon指数均无显著差异(P>0.05)。

图2 常用实验动物与人口腔菌群加权Unifrac距离(A1,A2)和未加权Unifrac距离(B1,B2)Fig.2 Weighted UniFrac distance (A1,A2) and unweighted UniFrac distance B1,B2) of oral flora in commonly used laboratory animals and humans
2.2.2 β多样性分析:β多样性研究群落之间菌落的相似性关系,图2(A1、B1)中一个点表示一个样品,样品之间的距离代表样品之间的相似度,距离越近,相似度越高。加权UniFrac距离在未加权的基础上增加了菌群的丰度。图2显示:除人之外,几个常用实验动物的样品间距离较近,相似度较高;无论加权和未加权Unifrac距离中,猴与人的距离相对较近(见图2中A2、B2),表明猴的口腔菌群相对于其他种类动物与人更相似,猴可能是研究人口腔菌群较适宜的模型动物。

图3 常用实验动物与人口腔菌群结构门水平分布Fig.3 Distribution of oral flora at phylum level in commonly used laboratory animals and humans
2.2.3口腔菌群结构组成:
2.2.3.1菌群结构门水平:图3显示:人、猪、犬、猴、兔、大鼠口腔菌群均以放线菌门(Actinobacillus)、拟杆菌门(Bacteroides)、变形菌门(Proteobacteria)和厚壁菌门(Firmicutes)为主,均占80%以上;人的口腔菌群中,放线菌门(Actinobacillus)、拟杆菌门(Bacteroides)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和梭杆菌门(Fusobacterium)五个门的细菌占主要地位,高达97.1%,在五种实验动物中,猴以上五个菌门的水平与人最相似;猴的梭杆菌门(Fusobacteria)比其它几种动物与人更相似;猪的变形菌门(Proteobacteria)显著高于人与其它几种动物;犬的螺旋体门(Spirochaetes)显著高于人和其它几种动物。
2.2.3.2菌群结构属水平:
从图4可以看出,人与不同种类实验动物的口腔菌群在属水平上均差异较大。人口腔中数量最多的是普氏菌属(Prevotella),这种菌属在猪、犬、猴、兔、鼠的口腔中均分布较少;猪、猴、兔、鼠的口腔菌群中均含有链球菌,猴口腔的链球菌含量最高;五种动物中,猴口腔菌群中梭菌属(Fusobacterium)、卟啉菌属(Porphyromonas)与人最相似。
2.2.4不同物种口腔菌群的统计学分析及生物标志物:
为进一步确定菌群种属的丰富度差异,采用在线统计工具LEfSe来确定人与5种动物的差异菌属,LDA值设置为4。结果表明:人的普氏菌属(Prevotella)(从门到属)、梭菌属(Fusobacterium)(从门到属)、奈瑟氏菌属(Neisseria)丰富度较高;猴的链球菌属(Streptococcus)(从门到属)、棒杆菌属(Corynebacterium)(从科到属)、巴斯德氏菌科(Pasteurellaceae)(从目到科)、毛螺旋菌科(Lachnospiraceae)、放线杆菌属(Actinobacillus)丰富度较高;猪的莫拉氏菌属(Moraxella)(从门到属)、金氏菌属(Kingella)丰富度较高;犬的梭菌目(Clostridiales)(从纲到目)、卟啉菌属(Porphyromonas)丰富度较高;兔的丛毛单胞菌科(Comamonadaceae)(从目到科)、鞘脂杆菌目(Sphingobacteriales)(从纲到目)、黄杆菌科(Flavobacteriaceae)(从纲到科)丰富度较高;鼠的罗氏菌属(Rothia)(从门到属)丰富度较高。
3 讨论
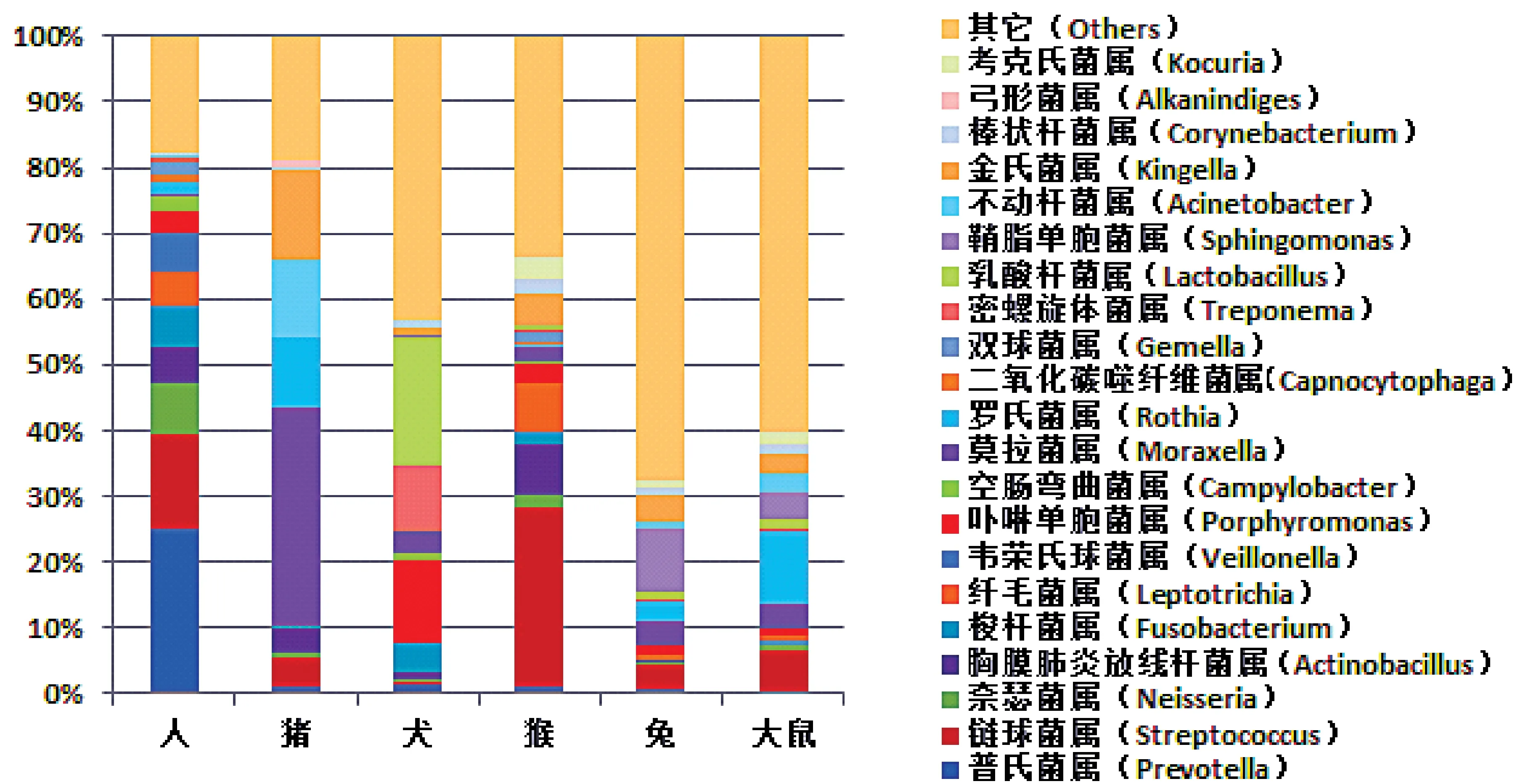
图4 常用实验动物与人口腔菌群结构属水平分布Fig.4 Distribution of oral flora at genera level in commonly used laboratory animals and humans
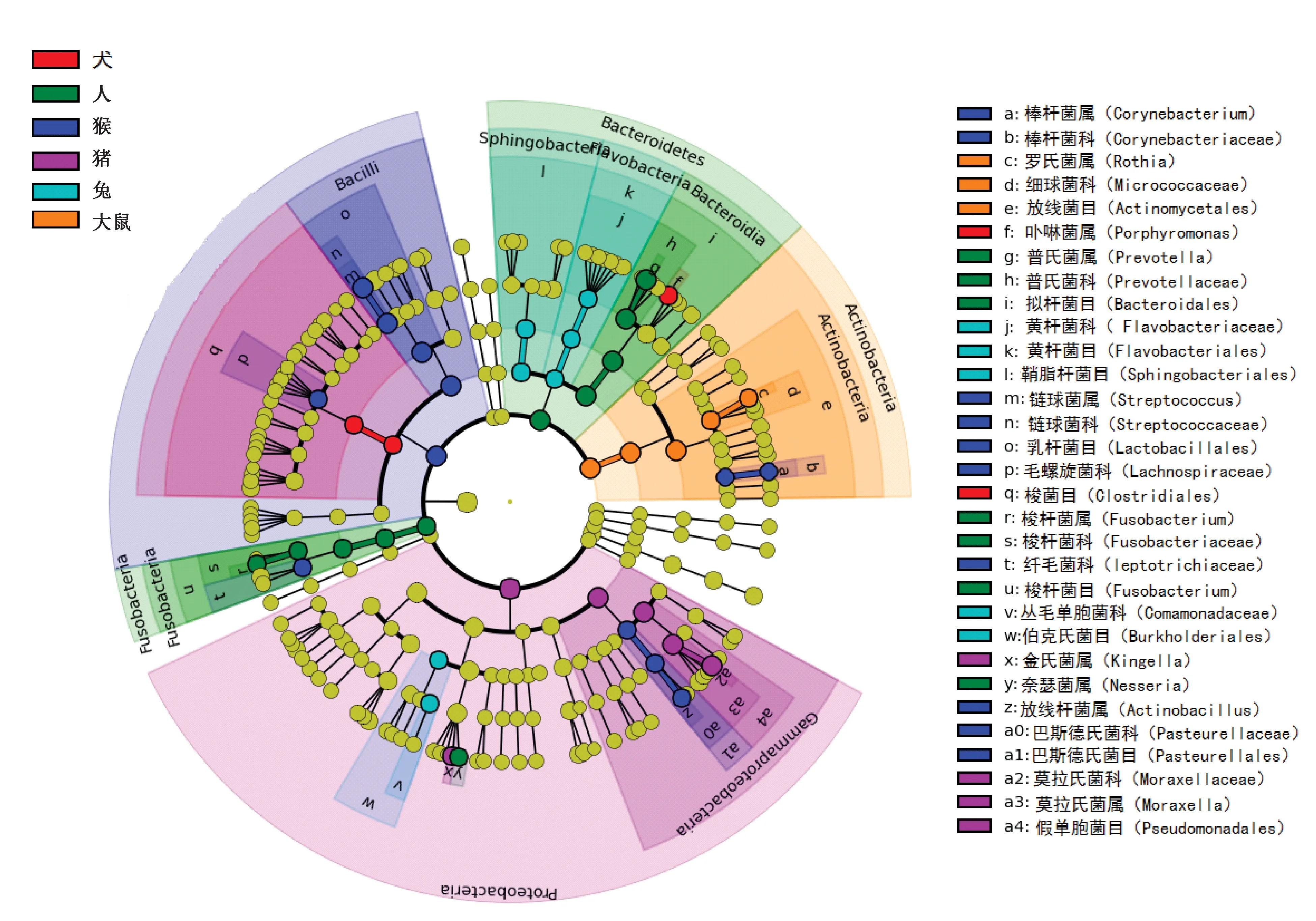
图5 常用实验动物与人口腔菌群Lefse结果进化分支图(LDA=4.0,P<0.05)Fig.5 LEfSe results of oral flora in commonly used laboratory animals and humans (LDA=4, P<0.05)
人与5种常用实验动物口腔菌群丰度差异显著(P<0.05),除了物种本身影响外,可能与彼此之间的饮食、环境等多种因素有关。菌群β多样性分析显示:猴与人的距离相对较近,表明猴的口腔菌群相对于其他种类动物与人更相似,提示猴可能是研究人口腔菌群较适宜的模型动物。人的口腔菌群中,放线菌门、拟杆菌门、变形菌门、厚壁菌门和梭杆菌门5个门的细菌占主要地位,高达97.1%,在五种实验动物中,猴以上5个菌门的水平与人最相似,这可能因为猴与人的亲缘关系最近,以及人工驯养繁殖的猴与人的饮食较相似有关;梭杆菌门是导致人口腔疾病的重要菌门,在5种动物中,猴与人的梭杆菌门水平最相似,提示猴可能是研究人与梭杆菌门细菌相关口腔疾病的较适宜模型;变形菌门包括很多病原菌,如变形杆菌、沙门氏菌、弧菌、螺杆菌等,与口腔感染性疾病密切相关,猪的变形菌门较其它几种动物丰富,提示猪可能是研究与变形菌门相关疾病的较适宜模型动物;螺旋体是人口腔正常菌群的一个组成部分,与牙周疾病的发生关系密切,在坏死性龈炎病变部位,可以发现大量的螺旋体,在慢性牙周炎的病损区也可发现菌斑中的螺旋体[12],在5种动物中,只有犬口腔的螺旋体门较为丰富,提示犬可能是研究与螺旋体门相关疾病的适宜模型动物。
人口腔中数量最多的是普氏菌属,是牙周病和牙周脓肿的致病菌[13],这种菌属在五种动物中均分布较少。梭菌属可与螺旋体混合感染,会引起急性溃疡性龈炎、急性坏死龈炎等,当人的炎症性牙周病变得严重时,此二类菌数大为增加,疾病减轻时又会减少,是牙周疾病治疗效果观察指标之一;卟啉菌属是口腔菌群中和牙周病相关的一个菌属[14],它可侵入人牙龈成纤维细胞,并且在相当浓度的抗生素作用下存活[15]。研究表明,它和类风湿关节炎也有一定的关联,类风湿关节炎症的患者普遍患有牙周疾病,并且使用抗生素的水平偏高[16];本研究中猴口腔的梭菌属、卟啉菌属比其他种类动物与人更相似,提示猴可能是研究与此菌属相关疾病的适宜模型动物。
目前,人体口腔菌群的研究较多,实验动物与人体之间正常口腔菌群的对比研究进行的很少;某一物种下的不同条件或刺激之间的对比研究较多,不同物种之间相同条件下的比较研究较少。与以往类似的研究相比,本研究的不同之处主要在于以下两点:以往的研究多采用2~3种实验动物,而本研究涵盖了医学研究中较常用的5种常用实验动物(猪、犬、猴、兔、大鼠),研究的动物种类最多;以往的研究多采用变性梯度凝胶电泳(DGGE)技术,本研究采用16Sr RNA高通量测序法,能够较为全面和准确的反映微生物群落结构,而DGGE仅能够反映有限的优势微生物类群,在技术水平上具有局限性。
[1]张存宝, 张雄. 口腔微生态研究进展 [J]. 中国乡村医药, 2012, 19(14):81-82.
[2]王鹏. 宏基因组法检测侵袭性牙周炎治疗前后菌斑微生物多样性 [D]. 山东大学, 2015.
[3]杨永华, 姚健. 分子生物学方法在微生物多样性研究中的应用 [J]. 生物多样性, 2000, 8(3): 337-342.
[4]周伟, 张东升. 口腔微生物多样性研究进展 [J]. 中国微生态学杂志, 2015, 27(6): 738-741.
[5]邓冠华, 查龙应, 张国霞, 等. 高通量16S rRNA标签测序法比较人与不同动物肠道微生物组多样性 [J]. 生态科学,2014, 33(05): 851-857.
[6]Zhou HW, Li DF, Tam NF, et al. BIPES, a cost-effective high-throughput method for assessing microbial diversity [J]. ISME J, 2011, 5(4): 741-749.
[7]Huse SM, Dethlefsen L, Huber JA, et al. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing [J]. PLoS Genet, 2008, 4(11): e1000255.
[8]Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities [J]. Appl Environ Microbiol, 2009, 75(23): 7537-7541.
[9]Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data [J]. Nat Methods, 2010, 7(5): 335-336.
[10]Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation [J]. Genome Biol, 2011, 12(6): R60.
[11]李岩涛, 李金陆, 杨圣辉, 等. 小型猪、大鼠及人口腔常见菌群的比较研究 [J]. 北京口腔医学, 2006, 14(03):153-156.
[12]吴娅菲, 杨美薷, 肖晓蓉, 等. 口腔菌群中螺旋体的分布[J]. 中国微生态学杂志, 1990, (1).69
[13]Tanaka S, Yoshida M, Murakami Y, et al. The relationship of Prevotella intermedia, Prevotella nigrescens and Prevotella melaninogenica in the supragingival plaque of children, caries and oral malodor [J]. J Clin Pediatr Dent, 2008, 32(3): 195-200.
[14]Naito M, Hirakawa H, Yamashita A, et al. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis [J]. DNA Res, 2008, 15(4): 215-225.
[15]Irshad M, van der Reijden WA, Crielaard W, et al. In vitro invasion and survival of Porphyromonas gingivalis in gingival fibroblasts; role of the capsule [J]. Arch Immunol Ther Exp (Warsz), 2012, 60(6): 469-476.
[16]Wegner N, Wait R, Sroka A, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis [J]. Arthritis Rheum, 2010, 62(9): 2662-2672.
Comparison of human and animal oral microbiota by Illumina sequencing of 16S rRNA tags
GU Dong-shu1, CHEN Bang-zhu1, JIANG Xia1, LIU Hai-yue2,Nashun Bayaer3, ZHOU Hong-wei4, GU Wei-wang1
(1. Laboratory Animal Center of Southern Medical University, Guangzhou 510515, China;2. Department of Environmental Health, School of Public Health and Tropical Medicine, Southern Medical University,Guangzhou 510515; 3. Guangdong Medical University, Dongguan 523808;4. Department of Laboratory Medicine of Zhujiang Hospital, Southern Medical University, Guangzhou 510282)
Objective To provide original reference data for oral ecosystem research, Tibet minipigs, beagle dogs, rhesus monkey, New Zealand white rabbits and Wistar rats were selected to study their respective characteristics of oral microbial mmunities and compared with normal data of humans. Methods Total DNA was extracted from the specimens of oral microbial communities of Tibet minipigs, beagle dogs, rhesus monkey, New Zealand white rabbits and Wistar rats, and used to amplify 16S rRNA V4 fragments with labeled universal primers. The diversity and structure of microbial communities from those animals were compared with that of humans using BIPES and QIIME analysis after Illumina sequencing of 16S rRNA V4 fragments. ResultsThe richness of the oral microbial communities of humans and the five species of laboratory animals was significantly different (P<0.05). Different species of animals have their own unique oral flora, among which the oral flora of the monkey is the most similar to that of humans. ConclusionsAmong the five species of laboratory animals, the oral microbial communities of rhesus monkeys and humans have highest similarity. Specifically, the Fusobacterium and Porphyromonas levels of rhesus monkeys is most similar to those of humans. Our findings indicate that rhesus monkeys may be suitable animal model for studies of human oral microbial communities. Tibet minipigs may be suitable animal model for Proteobacteria studies, while beagle dogs may be appropriate for modeling of diseases related to Spirochaetes.
Laboratory animal; Tibet minipig; Beagle dog; Rhesus monkey; New Zealand white rabbit; Wistar rat; Oral microbiota; 16S rRNA
广东省科技计划项目(编号:2013B030300040,2015A030302076);东莞松山湖高新区生物医药外包服务基地建设(编号:2012B011000004)。
顾东曙(1979-),女,硕士,专业:动物学。Email: 20595411@qq.com。
顾为望(1956-),男,教授,研究方向:实验动物培育、人类疾病动物模型制备与比较医学。Email: guww100@163.com。
研究报告
R-33
A
1671-7856(2016) 08-0096-07
10.3969.j.issn.1671-7856.2016.08.016
2016-05-02
猜你喜欢
中老年保健(2022年2期)2022-08-24 03:20:50
新民周刊(2022年27期)2022-08-01 07:04:49
传染病信息(2021年6期)2021-02-12 01:52:58
科学(2020年4期)2020-11-26 08:27:06
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
动物营养学报(2015年10期)2015-12-01 02:26:20
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
