
多孔纳米CoFe2O4的制备及其对高氯酸铵的热分解催化性能
2016-09-09 09:36:00熊文慧张文超俞春培沈瑞琪叶家海秦志春
物理化学学报 2016年8期
熊文慧 张文超 俞春培 沈瑞琪 程 佳 叶家海 秦志春
(南京理工大学化工学院,南京210094)
多孔纳米CoFe2O4的制备及其对高氯酸铵的热分解催化性能
熊文慧张文超*俞春培沈瑞琪程佳叶家海秦志春
(南京理工大学化工学院,南京210094)
采用胶晶模板法制备出具有三维多孔结构的纳米CoFe2O4。利用X射线衍射仪(XRD)、傅里叶变换红外(FT-IR)光谱仪、扫描电镜(SEM)、透射电镜(TEM)和N2吸附-脱附对样品的晶型和形貌结构等进行表征,采用差示扫描量热法(DSC)对比研究多孔纳米CoFe2O4和球形纳米CoFe2O4对高氯酸铵(AP)的热分解性能的影响,并考察这两种催化剂对AP催化热分解的动力学参数。结果显示,制备出的多孔纳米CoFe2O4样品具有典型的尖晶石结构,孔径约200 nm;比表面积明显高于40 nm球形CoFe2O4,达到55.646 m2·g-1。DSC测试结果表明:多孔纳米CoFe2O4的加入促进了AP的热分解,最高使AP的高温分解峰温降低91.46°C,能量释放最高达1120.88 J·g-1,是纯AP分解放热量的2.3倍;多孔纳米CoFe2O4具有较高的比表面积,能提高催化反应的接触面积,使AP的高温分解峰温度更低,反应活化能较小,从而表现出比球形纳米CoFe2O4更高的催化活性。此外,对多孔纳米CoFe2O4催化AP的热分解机理进行初步探索,纳米多孔催化剂对气态中间产物的作用促进了AP的热分解。
胶晶模板;多孔纳米CoFe2O4;高氯酸铵;热分解;催化
www.whxb.pku.edu.cn
1 引言
由于高氯酸铵(AP)具有较强的氧化特性,与固体推进剂组分有良好的相容性且受热分解能够产生大量的气体,而被应用于固体火箭推进剂中1,2。在AP系固体推进剂中,AP作为高能组分,一般占推进剂总质量的60%-90%,AP的活化能和热量释放等性质对推进剂的燃烧性能有重要影响3-5。从降低AP高温分解温度和提高AP的释放热量两个方面,来达到缩短推进剂的点火时间和提高推进剂的燃烧速率目的,是非常切实有效的6,7。
为了提高固体推进剂的燃烧性能,许多学者研究了催化剂对AP热分解性能的影响。其中,纳米金属氧化物由于具有高比表面积及高表面活性,常被用于固体推进剂中,如纳米α-Fe2O3、NiO、β-MnO2、Mn3O4、Co3O4、Cu2O等4,8-14。这些纳米金属氧化物对AP都有较高的催化活性,均能降低AP的热分解温度,提高AP分解反应释放的热量,对推进剂的燃烧性能有明显促进作用。而相对于单一金属氧化物,基于铁氧体的复合过渡金属氧化物由于具有较好的未填满的电子轨道,更容易接受电子,使电子转移加速,并且在复合金属氧化物的协同作用下,表现出比单一金属氧化物更优异的催化性能,从而引起了人们的关注5,15。CoFe2O4作为一种典型的尖晶石结构铁氧体,晶体表面存在大量的活性还原位点,其未填满的3d轨道能促进反应时的电子转移过程,最终使催化剂的催化效果增强16。然而,纳米催化剂粒子在制备和应用过程中容易团聚,会导致反应时与催化剂接触面积减小,从而影响催化剂性能的发挥17,18。很多研究者通过将纳米催化剂负载在碳纳米管和石墨烯等载体上,使催化剂的团聚问题得到改善,虽然在一定程度上使AP的热分解性能得到提升,但制备此类催化剂的过程相对复杂且容易出现其他杂质,使纳米催化剂难以发挥出最大的催化性能19,20。利用模板法制备的三维大孔结构材料,具有较大的比表面积和较高吸附性,能提高催化过程中的接触面积,增加活性位点,在一定程度上增强纳米催化剂的催化性能,并且制备工艺简单,结构可调,在催化领域中的潜在应用价值显著21,22。因此,将基于铁氧体的复合金属氧化物CoFe2O4与多孔纳米结构结合起来,利用两者独特的物化性质,制备出对AP具有高效催化分解性能的多孔纳米CoFe2O4是本文的重要工作内容。
为了研究多孔纳米复合金属氧化物对AP的催化分解性能影响,本文采用胶晶模板法制备一种具有多孔纳米结构的尖晶石型CoFe2O4,将其与AP混合,研究多孔纳米CoFe2O4对AP的催化效果,分析混合物中多孔纳米CoFe2O4的含量对AP的催化性能影响,探索多孔纳米CoFe2O4对AP的催化作用机理。
2 实验部分
2.1试剂和仪器
Co(NO3)2·6H2O、Fe(NO3)3·9H2O、甲醇、乙二醇:分析纯,国药集团化学试剂有限公司;40 nm球形CoFe2O4:分析纯,阿拉丁试剂有限公司;NH4ClO4:分析纯,上海麦克林生化科技有限公司。
采用德国Bruker AxS GmbH制造的D8 ADVANCE X射线衍射仪(XRD)对样品晶型进行表征,测试条件为Cu Kα辐射,工作电压为40 kV,工作电流为40 mA,扫描速率为8(°)·min-1,扫描范围10°-80°;采用Nicolet IS-10型傅里叶变换红外(FT-IR)光谱仪对样品结构进行表征,测试范围为525-4000 cm-1;采用HitachiS-4800型场发射扫描电镜(SEM)和日本电子(JEOL)JEM-2100型透射电子显微镜(TEM)对样品形貌进行表征;采用美国QUANTACHROME公司的Autosorb-IQ气体吸附分析仪测试样品的比表面积;采用瑞士Mettler Toledo生产的差示扫描量热仪(DSC)对AP的热分解过程进行测量,测试条件为:采用流速为15 mL·min-1的N2吹扫,在温度范围为50-500°C内以升温速率10°C·min-1程序升温。
2.2多孔纳米CoFe2O4薄膜的制备
采用无皂乳液聚合法制备聚苯乙烯(PS)微球乳液,通过垂直沉积法将单分散的聚苯乙烯微球组装成三维有序胶晶模板22。然后,在甲醇和乙二醇的混合溶液中,以Co(NO3)2·6H2O与Fe(NO3)3· 9H2O的摩尔比为1:2的比例配置前驱液。磁力搅拌一段时间后,将预热的胶晶模板放入前驱液中浸泡2 min,取出悬挂烘干。最后在马弗炉中以1°C· min-1的升温速率升温到500°C,保温3 h后冷却到室温即得到三维多孔结构的纳米CoFe2O4薄膜。
2.3多孔纳米CoFe2O4对AP催化分解性能研究
将制备的多孔纳米CoFe2O4薄膜样品剥离,将其分别以1%、2%、3%、4%、5%、6%、7%的质量分数(w)加入到AP中,加入一定量的丙酮不断搅拌并充分研磨,直至溶剂挥发,得到CoFe2O4/AP混合物。取约0.5 mg的混合物样品装于铝坩埚中,用差示扫描量热仪进行DSC分析。同时,为了研究纳米CoFe2O4的多孔结构与球形结构对AP的催化性能差异,在相同的测试条件下测试球形纳米CoFe2O4含量为5%时的AP热分解过程。作为空白对比研究,称取一定量的纯AP在同样的测试条件下进行DSC热分析,并通过分析AP的热分解温度、释放能量和反应活化能来衡量纳米CoFe2O4对AP的催化活性。
3 结果与讨论
3.1多孔纳米CoFe2O4的物相分析
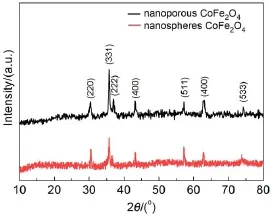
图1 多孔纳米CoFe2O4和球形纳米CoFe2O4的XRD图谱Fig.1 XRD patterns of nanoporous CoFe2O4and nanospheres CoFe2O4
图1是采用模板法制备的多孔纳米CoFe2O4和球形纳米CoFe2O4的XRD图谱。从图1可知,两种不同结构的纳米CoFe2O4分别在30.5°、35.4°、37.5°、43.0°、57.3°、63.0°、73.9°附近处存在明显的衍射峰,分别对应于CoFe2O4的标准图谱JCDPS 卡(No.22-1086)中的(220)、(331)、(222)、(400)、 (511)、(440)、(533)晶面,同时没有明显的杂质峰出现,说明通过模板法制备的多孔纳米CoFe2O4具有明显的立方尖晶石结构,得到的是晶化程度较高的多孔纳米CoFe2O4。图2是制备的多孔纳米CoFe2O4的红外光谱图。由于制备的样品中含有少量水分,图2中的曲线在波数为3384和1604 cm-1附近有较弱的吸收峰。此外,在525-600 cm-1之间出现了一个明显的谱带,这是位于586 cm-1的强吸收峰对应于CoFe2O4晶格中正八面体的特征收缩振动23。
3.2多孔纳米CoFe2O4的形貌结构分析
图3是制得的PS胶晶模板的SEM图和两种不同纳米结构CoFe2O4的SEM和TEM图。从图3(a)中可以看出,制备的PS微球表面光滑,粒径均匀,平均粒径约300 nm。通过垂直沉积组装的胶晶模板呈面心立方结构堆积,排列整齐有序。从整体上看,制备的PS胶晶模板在一定范围内能呈现出良好的有序性,存在的晶界和区域缺陷较少,可以作为一种周期性良好的多孔结构模板。图3(b)和图3(c)是利用模板法制备的多孔纳米CoFe2O4的SEM和TEM图片。配制的复合氧化物前驱体醇溶液在微球间隙间的毛细作用力下,填充到胶晶模板的间隙中。最后,通过煅烧除去PS胶晶模板得到多孔复合氧化物。这可以看作是一个PS胶晶模板的反向复制过程,PS微球被孔代替,仍呈面心立方结构紧密排列。每层骨架的球形孔都是相通的,孔壁厚在30-50 nm之间,孔径约200 nm,发生明显的收缩。图3(d)是球形纳米CoFe2O4的TEM图,粒径约40 nm,并发现其超声分散后仍团聚严重。因此,多孔纳米CoFe2O4在一定程度上可以体现出纳米催化剂在分散性问题上的优势。
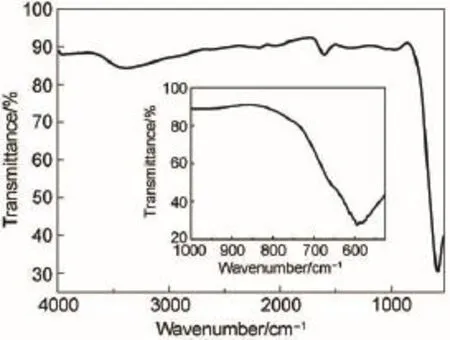
图2 多孔纳米CoFe2O4的FT-IR图谱Fig.2 FT-IR spectrum of nanoporous CoFe2O4

图3 (a)聚苯乙烯(PS)胶晶模板的SEM图片;(b)多孔纳米CoFe2O4的SEM图片;(c)多孔纳米CoFe2O4的TEM图片;(d)球形纳米CoFe2O4的TEM图片Fig.3 (a)SEM image of colloidal crystal template of polystyrene(PS);(b)SEM image of nanoporous CoFe2O4; (c)TEM image of nanoporous CoFe2O4;(d)TEM image of nanospheres CoFe2O4
为了研究多孔纳米CoFe2O4和球形纳米CoFe2O4样品的结构性质,分别对这两种样品进行氮气吸附-脱附实验,结果见图4(a)和4(b)。可以看出,两种纳米结构CoFe2O4样品的氮气吸附-脱附等温线均为Ⅲ型等温线,这种等温线会在大孔固体上发生弱的气-固相互作用时出现,并在相对压力在0.7-1.0之间出现H3型滞后环24-26。经Brunauer-Emmett-Teller(BET)方法计算出多孔纳米CoFe2O4的比表面积为55.646 m2·g-1,球形纳米CoFe2O4的比表面积为32.167 m2·g-1。显然,多孔纳米CoFe2O4样品比球形纳米的CoFe2O4比表面积更高,这也说明了多孔纳米结构可以减少纳米粒子的团聚12。
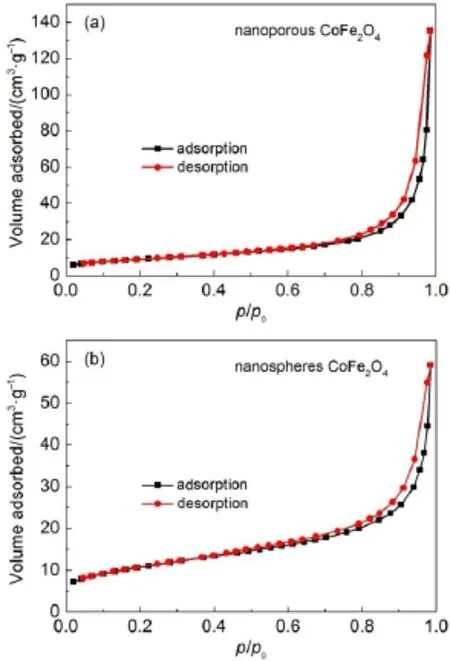
图4 (a)多孔纳米CoFe2O4和(b)球形纳米CoFe2O4的氮气吸附-脱附等温线Fig.4 Nitrogen adsorption-desorption isotherms for (a)nanoporous CoFe2O4and(b)nanospheres CoFe2O4
3.3多孔纳米CoFe2O4对AP的催化性能分析
图5为纯AP和含量为1%-7%(w)的多孔纳米CoFe2O4与AP混合后的DSC曲线。从图5(a)中可以看出,纯AP的热分解过程分三步进行。第一阶段是AP的相变吸热过程,晶体从斜方晶系转变为立方晶系,转变温度在240-250°C之间,纯AP样品的晶型转变吸热峰温度为245.94°C,与文献2,27,28报道的相一致。随着温度的升高,纯AP的分解分两阶段进行。当温度升高到300°C附近时进行低温分解,分解起始温度为274.56°C,低温分解放热峰温度为315.69°C。当温度增加到360°C左右时,AP开始高温分解,高温分解放热峰温度为413.89°C,完全分解生成HCl、H2O、Cl2、O2、NO、N2O和NO2等挥发性产物29,并放出热量488.27 J·g-1。

图5 不同含量的多孔纳米CoFe2O4与AP复合物的DSC曲线Fig.5 DSC curves of theAPdecomposition in the absence and presence of different blend ratios of nanoporous CoFe2O4
由图5可以看出:当加入多孔纳米CoFe2O4复合氧化物后,AP的热分解过程发生了明显变化。根据DSC曲线分析,多孔纳米CoFe2O4对AP受热分解过程中的晶型转变温度几乎没有影响,不同含量的多孔纳米CoFe2O4使AP的低温分解温度和高温分解温度均提前,分解反应放热量也大大提高,具体热分解反应数据如表1所示。当多孔纳米CoFe2O4含量为1%时,低温分解峰温度降低到257.14°C,高温分解峰温度降低到354.55°C,释放热量950.55 J·g-1。多孔纳米CoFe2O4的含量高于2%时,AP的低温分解峰消失,高温分解峰逐渐提前,峰温从350°C附近(图5(b,c))降低到约320°C(图5(g,h))。与纯AP相比,多孔纳米CoFe2O4最高可使AP的高温分解峰温降低91.46°C(含量为6%),分解反应热最高达1120.88 J·g-1(含量为2%),是纯AP分解反应热的2.3倍。可以看出,多孔纳米CoFe2O4对AP的分解有明显的促进作用,不仅降低AP分解反应温度,还提高了反应释放热量。然而,当多孔纳米CoFe2O4复合氧化物的含量增大到一定程度时,由于高温反应会出现催化剂比表面积下降,活性位点减少的现象,会使催化剂的活性降低4,12,30,31,AP的高温分解峰温降低的趋势也渐渐平稳,反应放热量也没有明显的增长趋势。
为了研究纳米复合氧化物的多孔结构对AP的催化热分解性能的影响,本文利用平均粒径为40 nm的球形CoFe2O4催化AP来进行对比研究。图6是两种不同纳米结构的CoFe2O4对AP的热分解DSC曲线。从图6中可以看出,当催化剂纳米CoFe2O4的质量分数均为5%时,两种纳米结构的CoFe2O4对AP都具有优异的催化效果。球形纳米CoFe2O4使AP的低温分解放热峰温度和高温分解放热峰温度分别提前了27.36和65.69°C。在分解过程中,AP的低温分解峰和高温分解峰融合,并释放热量910.18 J·g-1,是纯AP分解反应热的1.8倍。
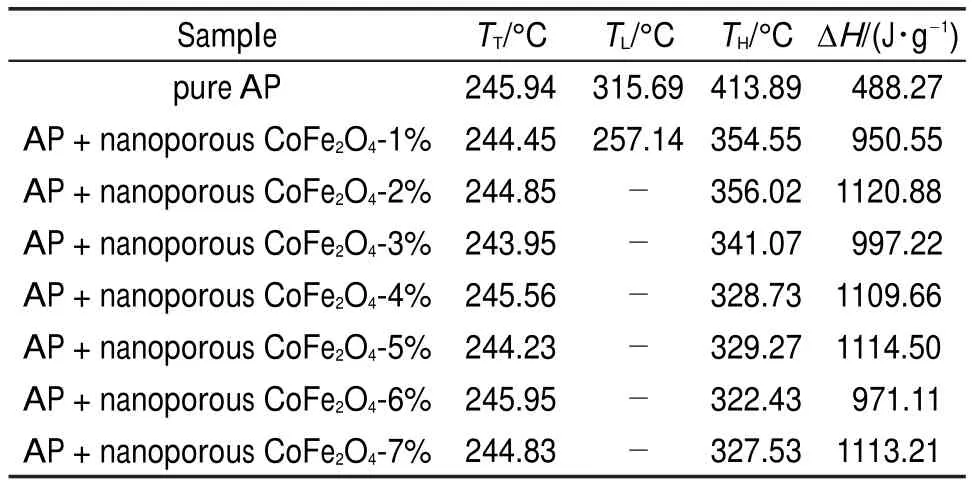
表1 不同组分的CoFe2O4/AP热分解DSC数据Table 1 Data of theAPdecomposition in the absence and presence of different blend ratios of nanoporous CoFe2O4
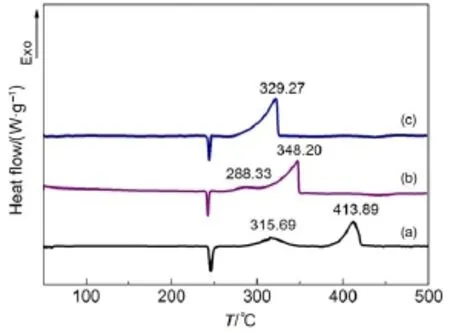
图6 多孔纳米与球形纳米结构的CoFe2O4与AP复合物的DSC曲线Fig.6 DSC curves of theAPdecomposition in the presence of nanoporous and nanospheres CoFe2O4
在动力学研究中,对两种不同结构的纳米CoFe2O4催化AP热分解的动力学参数进行对比。在催化剂含量(质量分数为5%)相同的条件下,升温速率分别为2、5、10和20°C·min-1,对AP进行DSC分析(图7),并利用Kissinger法对AP分解反应的活化能等动力学参数进行计算32,结果如表2。从表2中可以看出,在加入5%含量的纳米CoFe2O4后,球形纳米CoFe2O4催化AP分解的活化能为94.64 kJ·mol-1,多孔纳米CoFe2O4催化AP分解的活化能为74.02 kJ·mol-1。显然,多孔纳米CoFe2O4使AP热分解的活化能更低,从这一方面也显示出比球形纳米CoFe2O4更佳的催化活性7。
因此,与球形纳米结构的CoFe2O4热分解参数相比,多孔纳米结构的CoFe2O4对AP的催化效果明显优于球形结构。由于AP热分解的中间气态产物在催化剂表面进行反应,具有较高比表面积的多孔CoFe2O4会增强气态分子在催化剂表面吸附和解吸作用,使AP的高温分解活化能低于球形结构催化剂,高温分解温度更低,从而表现出优异的催化性能4。
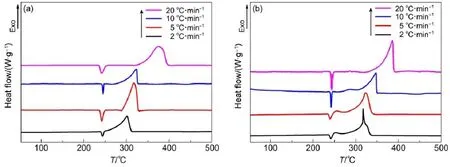
图7 多孔纳米与球形纳米结构的CoFe2O4与AP复合物以不同升温速率的DSC曲线Fig.7 DSC curves of theAPdecomposition in the presence of nanoporous and nanospheres CoFe2O4at given heating rates

表2 多孔纳米与球形纳米结构的CoFe2O4与AP复合物的动力学参数(Kissinger法)Table 2 Kinetic parameters of theAPdecomposition in the presence of nanoporous and nanospheres CoFe2O4based on Kissinger method
3.4多孔纳米CoFe2O4对AP的催化机理分析
AP的分解过程是一个固-气多相反应过程15。低温时,AP分解形成少量的中间产物,部分离解和升华形成NH3和HClO4:
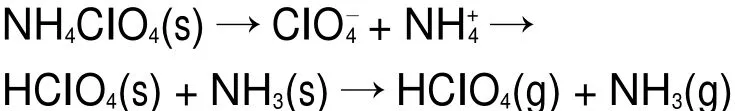
然而,中间产物NH3和HClO4间的反应并不完全,未反应的NH3会吸附在AP表面。当AP表面完全被NH3包覆时,AP的分解反应停止,反应过程如图8所示。当温度继续升高时,吸附AP表面的NH3与HClO4重新开始反应,最终生成挥发性产物18。
当加入纳米CoFe2O4作为催化剂时,复合过渡金属氧化物中存在处于未被填满状态的轨道,容易接受ClO-4电子,并转移到催化剂表面,使N接受电子随后分解,形成NH3,Cl转变成HClO4。此时,CoFe2O4晶格表面存在的Fe2+/Fe3+、Co2+/Co3+和Fe3+/Co2+作为活性还原位点,能引发电子迁移,并为催化反应提供了电子转移通道。反应过程如下16,30:
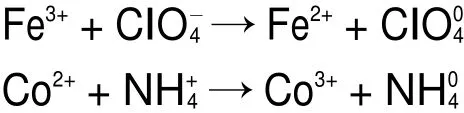
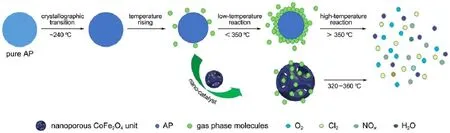
图8 纯AP和AP/多孔纳米CoFe2O4复合物分解过程示意图Fig.8 Schematic diagram of the thermalAPdecomposition process in the absence and presence of nanoporous CoFe2O4
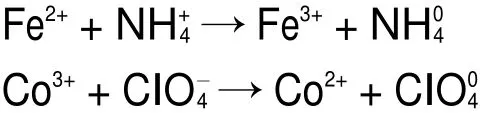
此外,由于多孔纳米复合金属氧化物具有高比表面积和高吸附性,AP受热分解形成的气态中间产物吸附在多孔纳米CoFe2O4的表面(如图8所示),使催化反应的接触面积增大,活性位点增加,反应会持续发生18,33,34。最终使吸附在多孔纳米CoFe2O4表面上的气态中间产物在活性位点上进行电子转移后解吸,脱离孔壁,生成HCl、H2O、Cl2、O2、NO、N2O和NO2等最终产物16,35。因此,表现出比球形纳米CoFe2O4更优异的催化性能。
4 结论
(1)通过胶晶模板法制备出具有三维多孔结构的纳米CoFe2O4,其孔径约200 nm,孔壁厚在30-50 nm之间。氮气吸附-脱附实验结果表明,多孔纳米CoFe2O4的比表面积明显高于40 nm球形CoFe2O4,达到55.646 m2·g-1。
(2)DSC数据分析表明,多孔纳米CoFe2O4对AP的低温分解和高温分解阶段都有明显的促进作用。当加入质量分数为1%的多孔纳米CoFe2O4时,AP的低温分解峰温度降低了58.55°C,高温分解峰温度降低了59.34°C。随着加入的多孔纳米CoFe2O4质量分数的增加,AP低温分解峰逐渐消失,高温分解峰提前,最高使AP高温分解峰温度提前91.46°C(质量分数为6%)。同时,催化AP分解的热量释放最高提高到1120.88 J·g-1(质量分数为2%),是纯AP分解反应热的2.3倍。
(3)当催化剂含量均为5%时,由于多孔催化剂的高比表面积和高吸附性,促进了气相中间产物在催化剂表面上的反应,使多孔纳米CoFe2O4催化AP的高温分解峰温度降低,反应释放热量增加,催化AP分解的活化能低于球形纳米CoFe2O4,显示出多孔结构的纳米CoFe2O4对AP热分解的高催化性能。这一结果凸显出多孔纳米结构金属复合氧化物作为催化剂的显著优越性,对其他催化领域也有巨大的潜在应用价值。
References
(1)Boldyrev,V.Thermochim.Acta 2006,443(1),1.doi:10.1016/j. tca.2005.11.038
(2)Bircumshaw,L.;Newman,B.H.Proc.R.Soc.London,Ser.A 1954,227(1168),115.doi:10.1098/rspa.1954.0284
(3)Chen,L.J.;Li,L.P.;Li,G.S.J.Alloy.Compd.2008,464,532. doi:10.1016/j.jallcom.2007.10.058
(4)Hosseini,S.G.;Ahmadi,R.;Ghavi,A.;Kashi,A.Powder Technol.2015,278,316.doi:10.1016/j.powtec.2015.03.032
(5)Alizadeh-Gheshlaghi,E.;Shaabani,B.;Khodayari,A.;Azizian-Kalandaragh,Y.;Rahimi,R.Powder Technol.2012,217,330. doi:10.1016/j.powtec.2011.10.045
(6)Yu,Z.X.;Chen,L.F.;Lu,L.D.;Yang,X.J.;Wang,X.Chin.J. Catal.2009,30(1),19.[余宗学,陈莉芬,陆路德,杨绪杰,王信.催化学报,2009,30(1),19.]doi:10.1016/S1872-2067(08) 60087-X
(7)Vyazovkin,S.;Wight,C.A.Chem.Mater.1999,11(11),3386. doi:10.1021/cm9904382
(8)Chaturvedi,S.;Dave,P.N.J.Saudi Chem.Soc.2013,17(2), 135.doi:10.1016/j.jscs.2011.05.009
(9)Kapoor,I.P.S.;Srivastava,P.;Singh,G.Propell.Explos.Pyrot. 2009,34(4),351.doi:10.1002/prep.200800025
(10)Sharma,J.;Srivastava,P.;Singh,G.;Akhtar,M.S.;Ameen,S. Ceram.Int.2015,41(1),1573.doi:10.1016/j. ceramint.2014.09.093
(11)Chandru,R.A.;Patra,S.;Oommen,C.;Munichandraiah,N.; Raghunandan,B.J.Mater.Chem.2012,22(14),6536. doi:10.1039/C2JM16169A
(12)Li,N.;Geng,Z.F.;Cao,M.H.;Ren,L.;Zhao,X.Y.;Liu,B.; Tian,Y.;Hu,C.W.Carbon 2013,54,124.doi:10.1016/j. carbon.2012.11.009
(13)Zhao,Y.J.;Xu,X.M.;Zhao,Y.Z.;Zhou,H.P.;Li,J.B.;Jin,H. B.J.Alloy.Compd.2016,654,523.doi:10.1016/j. jallcom.2015.09.145
(14)Luo,X.L.;Yang,D.S.;Yuan,C.L.;Luo,X.M.;Chen,Y.S. Acta Phys.-Chim.Sin.2014,30(3),520.[罗小林,杨得锁,原春兰,罗旭梅,陈亚芍.物理化学学报,2014,30(3),520.] doi:10.3866/PKU.WHXB201401061
(15)Wang,Y.P.;Xia,X.Y.;Zhu,J.W.;Li,Y.;Wang,X.;Hu,X.D. Combust.Sci.Technol.2010,183(2),154.doi:10.1080/ 00102202.2010.507561
(16)Said,A.A.;Al-Qasmi,R.Thermochim.Acta 1996,275(1),83. doi:10.1016/0040-6031(95)02721-1
(17)Silva,J.B.;Diniz,C.F.;Lago,R.M.;Mohallem,N.D.J.Non-Cryst.Solids 2004,348,201.doi:10.1016/j. jnoncrysol.2004.08.169
(18)Lan,Y.F.;Jin,M.M.;Luo,Y.J.J.Sol-Gel Sci.Technol.2015, 74(1),161.doi:10.1007/s10971-014-3590-3
(19)Xu,M.W.;Wang,F.;Zhang,Y.;Yang,S.;Zhao,M.S.;Song,X. P.Nanoscale 2013,5(17),8067.doi:10.1039/C3NR02538A
(20)Wang,Z.M.;Xu,C.L.;Gao,G.Q.;Li,X.RSC Adv.2014,4 (26),13644.doi:10.1039/c3ra47721e
(21)Kim,W.;Nair,S.Chem.Eng.Sci.2013,104,908.doi:10.1016/ j.ces.2013.09.047
(22)Zhang,W.C.;Yin,B.Q.;Shen,R.Q.;Ye,J.H.;Thomas,J.A.; Chao,Y.M.ACS Appl.Mater.Interfaces 2013,5(2),239. doi:10.1021/am302815y
(23)Yavuz,Ö.;Ram,M.K.;Aldissi,M.;Poddar,P.;Hariharan,S. J.Mater.Chem.2005,15(7),810.doi:10.1039/B408165J
(24)Thommes,M.Chem.Ing.Tech.2010,82(7),1059.doi:10.1002/ cite.201000064
(25)Kruk,M.;Jaroniec,M.Chem.Mater.2001,13(10),3169. doi:10.1021/cm0101069
(26)Wang,Y.L.;Zhao,C.H.;Fu,W.B.;Zhang,Z.M.;Zhang,M. X.;Zhou,J.Y.;Pan,X.J.;Xie,E.Q.J.Alloy.Compd.2016, 668,1.doi:10.1016/j.jallcom.2016.01.212
(27)Campos,E.A.;Fernandes,M.T.C.;Kawachi,E.Y.;de Oliveira,J.I.S.;Dutra,R.D.L.Propell.Explos.Pyrot.2015, 40(6),860.doi:10.1002/prep.201500115
(28)Zhou,L.M.;Liu,H.Y.;Li,F.S.Acta Phys.-Chim.Sin.2006, 22(5),627.[周龙梅,刘宏英,李凤生.物理化学学报,2006,22 (5),627.]doi:10.3866/PKU.WHXB20060521
(29)Jacobs,P.W.M.;Whitehead,H.Chem.Rev.1969,69(4),551. doi:10.1021/cr60260a005
(30)Liu,T.;Wang,L.S.;Yang,P.;Hu,B.Y.Mater.Lett.2008,62 (24),4056.doi:10.1016/j.matlet.2008.04.081
(31)Yi,P.;Jiang,X.H.;Zou,M.;Lu,L.D.;Wang,X.Chin.J.Inorg. Chem.2014,30(1),185.[尹萍,江晓红,邹敏,陆路德,汪信.无机化学学报,2014,30(1),185.]doi:10.11862/ CJIC.2014.076
(32)Morisaki,S.;Komamiya,K.Thermochim.Acta 1975,12(3), 239.doi:10.1016/0040-6031(75)85037-4
(33)Zeng,G.Y.;Yu,W.F.;Nie,F.D.;Huang,H.;Xia,Y.X.;Lv,C. X.Initiators Pyrotech.2007,5,16.[曾贵玉,郁卫飞,聂福德,黄辉,夏云霞,吕春绪.火工品,2007,5,16.]doi:10.3969/j. issn.1003-1480.2007.05.005
(34)Wei,Y.C.;Zhao,Z.;Jin,B.F.;Yu,X.H.;Jiao,J.P.;Li,K.X.; Liu,J.Catal.Today 2015,251,103.doi:10.1016/j. cattod.2014.08.034
(35)Liu,D.;Lu,Y.;Tan,H.Q.;Chen,W.L.;Zhang,Z.M.;Li,Y.G.; Wang,E.B.Chem.Commun.2013,49(35),3673.doi:10.1039/ c3cc40990b
Preparation of Nanoporous CoFe2O4and Its Catalytic Performance during the Thermal Decomposition of Ammonium Perchlorate
XIONG Wen-HuiZHANG Wen-Chao*YU Chun-PeiSHEN Rui-Qi CHENG JiaYE Jia-HaiQIN Zhi-Chun
(School of Chemical Engineering,Nanjing University of Science and Technology,Nanjing 210094,P.R.China)
Three-dimensional,nanoporous CoFe2O4catalysts were synthesized,employing a colloidal crystal template method.X-ray diffraction(XRD),Fourier transform infrared(FT-IR)spectroscopy,scanning electron microscopy(SEM),transmission electron microscopy(TEM),and N2adsorption-desorption were subsequently used to characterize the crystal structures and morphologies of the samples.The catalytic activities of nanoporous CoFe2O4and CoFe2O4nanospheres during the thermal decomposition of ammonium perchlorate (AP)were also investigated by differential scanning calorimetry(DSC).The results show that the spinel framework of these materials has an ordered open network of pores averaging 200 nm in diameter.The specific surface area of the nanoporous CoFe2O4was 55.646 m2·g-1,a value that was higher than that of the nanosphere material.DSC analysis indicates that the catalytic activity of the nanoporous CoFe2O4is superior to that of the spherical material during the thermal decomposition of AP,and that the nanoporous catalyst makes the peak temperature of high temperature decomposition decrease by 91.46°C.The heat release from the AP in the presence of nanoporous CoFe2O4(1120.88 J·g-1)is 2.3 times that obtained frompureAP.Both the higher specific surface area and greater quantity of active reduction sites on the nanoporous CoFe2O4relative to the nanospherematerial act to reduce the activation energy during theAP decomposition process.Based on the results of this work,a possible catalytic mechanismfor the thermal decomposition ofAPover nanoporous CoFe2O4is proposed, in which gaseous intermediates play an important role.
Colloidal crystal template;Nanoporous CoFe2O4;Ammonium perchlorate;Thermal decomposition;Catalysis
February 1,2016;Revised:May 11,2016;Published on Web:May 12,2016.
O643
10.3866/PKU.WHXB201605121
*Corresponding author.Email:zhangwenchao303@aliyun.com;Tel:+86-25-84315515.
The project was supported by the National Natural Science Foundation of China(51576101),Natural Science Foundation of Jiangsu Province,China (BK20141399),and Fundamental Research Funds for the Central Universities,China(30915012101).
国家自然科学基金(51576101),江苏省自然科学基金(BK20141399)及中央高校基本科研业务费专项资金(30915012101)资助项目
©Editorial office ofActa Physico-Chimica Sinica
[Article]
猜你喜欢
今日农业(2022年16期)2022-11-09 23:18:44
今日农业(2022年15期)2022-09-20 06:55:48
环球时报(2022-06-20)2022-06-20 17:06:23
陶瓷学报(2020年6期)2021-01-26 00:37:56
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
中学生数理化·中考版(2018年11期)2019-01-31 06:18:06
教学考试(高考化学)(2018年5期)2018-12-06 07:21:56
基层中医药(2018年8期)2018-11-10 05:32:06
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
