
4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑的合成和表征
2016-09-06 07:37毛桢东高鹏杰于秀丽李惠萍郑州大学化工与能源学院河南郑州450001
河南化工 2016年4期
毛桢东,王 磊,高鹏杰,于秀丽,李惠萍(郑州大学化工与能源学院,河南郑州 450001)
·开发与研究·
4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑的合成和表征
毛桢东,王磊,高鹏杰,于秀丽,李惠萍
(郑州大学化工与能源学院,河南郑州450001)
摘要:以氯化苄为原料,通过叠氮取代生成苄基叠氮(Ⅰ),然后利用点击化学法合成1-苄基-3-羟甲基-1H -1,2,3-三氮唑(Ⅱ),其次利用威廉森反应将(Ⅱ)接枝在γ-氯丙基三甲氧基硅烷上得到1-苄基-4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1,2,3-三氮唑(Ⅲ),最后脱除苄基得到4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑(Ⅳ),可用于制备燃料电池质子交换膜。用FT-IR、1H-NMR以及13C-NMR对产物的结构进行了表征,结果证实了合成路线的可行性。采用单因素实验和正交试验考察了反应温度、反应时间、反应物料物质的量比和催化剂用量等,在最佳工艺条件下得到每一步反应的平均收率分别为83.89%、92.60%、90.56%、71.39%。
关键词:点击化学;4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑;质子交换膜
商业化全氟磺酸膜,质子传递主要依赖于外部水分子,工作温度较低(80℃左右),导致铂催化剂一氧化碳中毒,引发电池内部水、热管理困难等问题[1-2]。因此,开发以非水质子载体为基础的新型质子交换膜备受关注[3-4]。文献报道,1H-1,2,3-三氮唑化合物具有较好的质子传导能力和电化学稳定性,三氮唑环上的H原子可以通过异构化分布于3个氮原子上,使其具有原生质子能力。因此,将其用于质子交换膜是可选择的方案之一[5-8]。
本研究首先由氯化苄经叠氮取代获得苄基叠氮(Ⅰ),产物(Ⅰ)再与丙炔醇合成1-苄基-4-羟甲基-1H-1,2,3-三氮唑(Ⅱ),接着将其接枝在硅烷偶联剂上得到1-苄基-4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1,2,3-三氮唑(Ⅲ),最后把保护基苄基脱除得到可用于制备质子交换膜的4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑(Ⅳ)。合成路线如下:
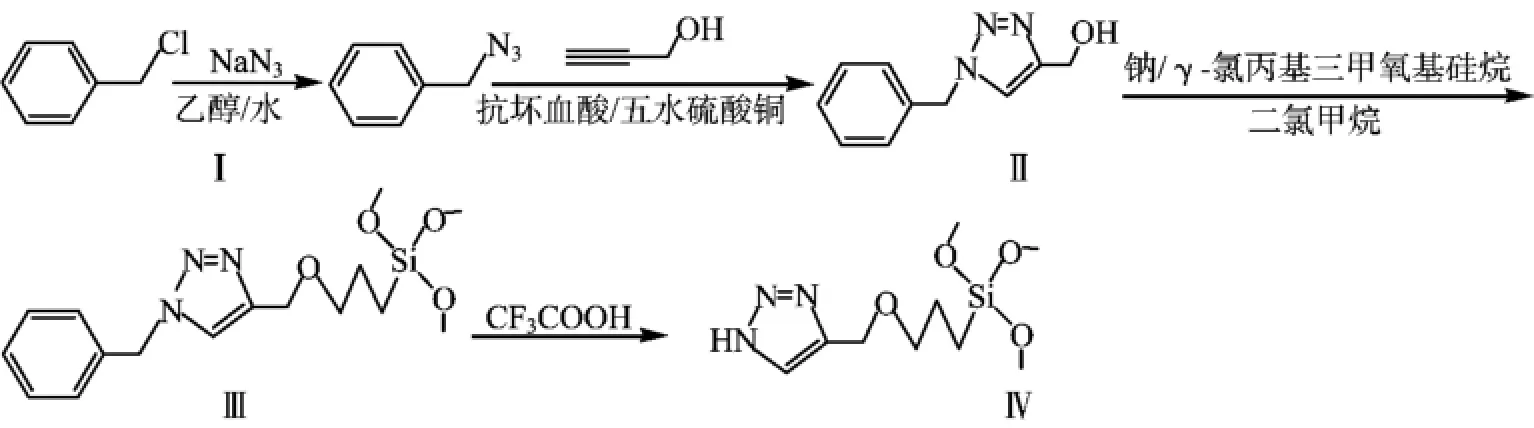
1 实验部分
1.1试剂与仪器
苄基氯:AR,国药集团化学试剂有限公司;叠氮钠:AR,天津凯通化学试剂有限公司;无水乙醇、无水甲醇:均为AR,天津市风船化学试剂科技有限公司;炔丙醇:AR,郑州鑫顺电镀公司;金属钠:AR,天津市福晨化学试剂厂;五水硫酸铜、四氢呋喃、抗坏血酸、γ-氯丙基三甲氧基硅烷、无水硫酸镁、二氯甲烷:均为AR,天津市科密欧试剂有限公司;水为实验室自制二次蒸馏水。
IR-200型傅里叶红外变换光谱仪,美国热电公司;DPX-400核磁共振仪,瑞士布鲁克公司。
1.2合成步骤
1.2.1苄基叠氮(Ⅰ)的合成
在100 mL三口烧瓶中,加入2.53 g(0.02 mol)苄基氯、1.625 g(0.025 mol)叠氮钠、15 mL无水乙醇、4mL水,75℃恒温磁力搅拌下回流反应6 h。反应结束后,将反应液抽滤以除去过量叠氮钠,滤液经旋蒸除去溶剂乙醇,在釜液中加入20 mL二氯甲烷和20 mL蒸馏水,用分液漏斗萃取得到有机相,水相用10 mL二氯甲烷萃取两次,合并有机层,加入适量无水硫酸镁干燥24 h,抽滤后将滤液旋蒸除去二氯甲烷,得到淡黄色油状液体即苄基叠氮,收率为83.89%。FT-IR(KBr),ν/cm-1:1600~1 449(苯环),2 096(-N3)。1H-NMR(CDCl3,400 MHz),δ(ppm):4.38(s,2H),7.40~7.41(m,3H),7.42~7.46(m,2H)。13C-NMR(CDCl3,400 MHz),δ(ppm):54.83,128.27,128.35,128.88,135.40。
1.2.21-苄基-3-羟甲基-1H-1,2,3-三氮唑(Ⅱ)的合成
在100 mL三口烧瓶中,加入苄基叠氮2.66 g (0.02 mol)、炔丙醇1.344 g(0.024 mol)、抗坏血酸0.704 g(0.004 mol)、五水硫酸铜0.1 g(0.000 4 mol)、四氢呋喃20 mL、蒸馏水5 mL,63℃恒温磁力搅拌下回流反应24 h,反应结束后经旋蒸除去溶剂四氢呋喃,在釜液中加入20 mL二氯甲烷和20 mL蒸馏水,用分液漏斗萃取得到有机相,水相用10 mL二氯甲烷萃取两次,合并有机层,加入适量无水硫酸镁干燥24 h,抽滤后将滤液旋蒸除去二氯甲烷,得到黄色凝胶状物质即为(Ⅱ),收率为92.60%。FTIR(KBr),ν/cm-1:1 017(三氮唑),3 266(—OH)。1H-NMR(CDCl3,400 MHz),δ(ppm):3.51 (brs,—OH),4.73(brs,2H),5.48(s,2H),7.24~7.34(m,5H),7.47(brs,2H)。13C-NMR(CDCl3,400 MHz),δ(ppm):54.22,56.16,122.00,128.14,128.80,129.13,134.49,148.61。
1.2.31-苄基-4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1,2,3-三氮唑(Ⅲ)的合成
在100 mL三口烧瓶中,加入γ-氯丙基三甲氧基硅烷1.89 g(0.01 mol)、金属钠0.345 g(0.015 mol)、1-苄基-3-羟甲基-1H-1,2,3-三氮唑(Ⅱ)1.985 g(0.01 mol)、二氯甲烷20 mL,在40℃恒温磁力搅拌下回流反应12 h,反应结束后,将反应液转移至50 mL烧杯中,把过量小钠块投入大量水中,烧杯中溶液经旋蒸后除去溶剂二氯甲烷,在釜液中加入20 mL二氯甲烷和20 mL蒸馏水,萃取得到有机相,水相用10 mL二氯甲烷萃取两次,合并有机层,加入适量无水硫酸镁干燥24 h,抽滤后经旋蒸除去二氯甲烷,得到黄绿色凝胶状物质即1-苄基-4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1,2,3-三氮唑(Ⅲ),收率为90.56%。FT-IR (KBr),ν/cm-1:1033(三氮唑),1120(Si—O),1201(C—O—C)。1H-NMR(CDCl3,400MHz),δ(ppm):0.77~0.83(m,2H),1.88~1.93(m,2H),3.54~3.60(t,9H),4.72(s,2H),4.79~4.83(s,2H),5.52~5.54(m,2H),7.28~7.30(m,2H),7.37~7.39(m,2H),7.49(s,H)。13C-NMR (CDCl3,400 MHz),δ(ppm):8.09,26.29,47.37,50.34,54.17,61.16,122.57,128.15,128.79,134.52,145.08。
1.2.44-{[3-(三甲氧基硅烷基)]甲基}-1H -1,2,3-三氮唑(Ⅳ)的合成
100 mL三口烧瓶中加入1-苄基-4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1,2,3-三氮唑(Ⅲ)3.51 g(0.01 mol)、三氟乙酸20 mL,72℃恒温反应为36 h。反应结束后,将反应液冷却至室温,经旋蒸除去溶剂三氟乙酸,在釜液中加入20 mL二氯甲烷和20 mL蒸馏水,用分液漏斗萃取得到有机相,水相用10 mL的二氯甲烷萃取两次,合并有机层,加入适量无水硫酸镁干燥24 h,抽滤后将滤液旋蒸除去二氯甲烷,得到红棕色凝胶状物质即为4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑(Ⅳ),收率为71.39%。FT-IR (KBr),ν/cm-1:1053(三氮唑),1151(Si—O),1 201(C—O—C)1770(N—H)。1H-NMR(CDCl3,400 MHz),δ(ppm):0.81~0.85(s,2H),1.88(s,1H),2.93~3.50(t,9H),3.56(m,2H),4.79(s,2H),7.45(s,1H),8.06(s,1H)。13C-NMR(CDCl3,4 00MHz),δ(ppm):9.35,26.28,47.05,54.48,58.04,128.27,133.73。
2 结果与讨论
2.1苄基叠氮(Ⅰ)的合成
通过单因素实验初步确定实验范围,在此基础上选取四个因素:A为n(叠氮钠)∶n(氯化苄),B为无水乙醇乙醇用量,C为反应时间,D为反应温度,且每个因素各选取三个水平进行正交试验设计。正交因素水平设计和实验结果如表1、表2所示。
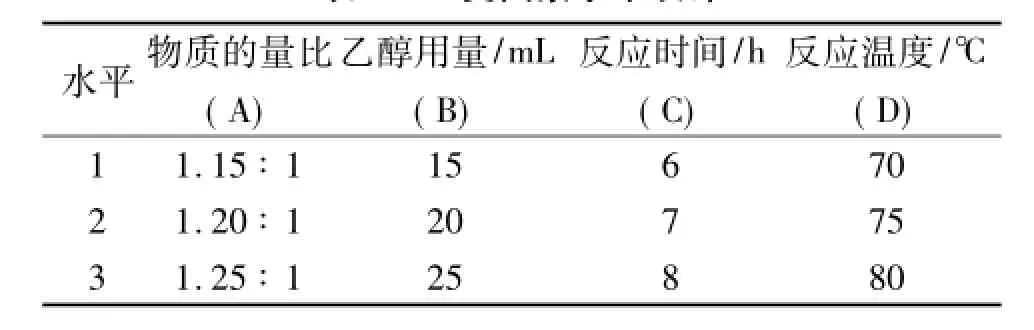
表1 正交因素水平设计
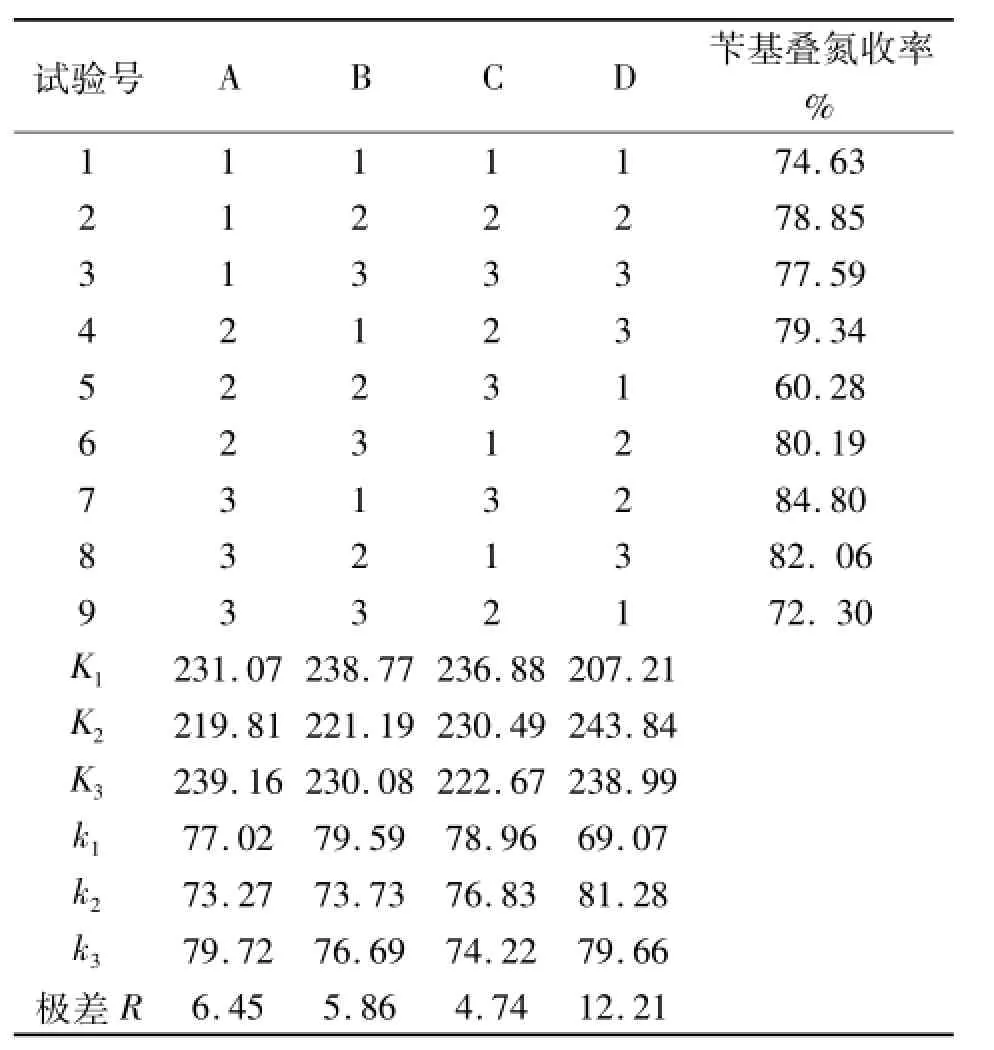
表2 正交试验结果
正交试验结果分析:根据极差R值大小,可判断对苄基叠氮收率影响由大到小顺序排列依次为:反应温度>反应物物质的量比>乙醇用量>反应时间;取每个因素中k的最大值,可确定正交试验最优方案为A3B1C1D2,即为n(叠氮钠)∶n(氯化苄)= 1.25∶1,无水乙醇用量15 mL,反应时间为6 h,反应温度为75℃。在此条件下做平行试验四次结果分别为84.06%、83.88%、84.71%、82.91%,平均收率为83.89%。
2.21-苄基-3-羟甲基-1H-1,2,3-三氮唑(Ⅱ)的合成
2.2.1四氢呋喃用量对收率的影响
在100 mL三口烧瓶中加入2.66 g苄基叠氮、1.344 g丙炔醇、0.352 g抗坏血酸、0.10 g CuSO4· 5H2O,加入5 mL蒸馏水,改变四氢呋喃的用量,温度60℃恒温反应24 h。实验结果如表3所示,增加四氢呋喃的用量,收率随之增大,当四氢呋喃用量为20 mL时有最大收率;继续增加四氢呋喃用量时,各反应物以及催化剂会被稀释,收率反而下降。由此可确定本实验最佳四氢呋喃用量为20 mL。

表3 四氢呋喃用量对收率的影响
2.2.2反应温度对收率的影响
在100 mL三口烧瓶中加入2.66 g苄基叠氮、1.344 g丙炔醇、0.352 g抗坏血酸、0.10 g CuSO4· 5H2O,加入5 mL蒸馏水和20 mL四氢呋喃,不同温度反应24 h。实验结果如表4所示,反应温度上升产物收率上升,在70℃达到最高收率69.78%;继续升温时,由于四氢呋喃与水在63℃附近形成共沸体系,溶液温度维持在共沸温度不再继续上升,因此设定63℃为最佳反应温度。

表4 反应温度对收率的影响
2.2.3反应时间对收率的影响
在100 mL三口烧瓶中加入2.66 g苄基叠氮、1.344 g丙炔醇、0.352 g抗坏血酸、0.10 g CuSO4· 5H2O,加入5 mL蒸馏水和20 mL四氢呋喃,改变反应时间,在63℃下恒温反应。实验结果如表5所示,延长反应时间,产物收率增加,24 h时得到最大收率77.82%;之后继续反应,产物收率基本不变,因此可得到24 h为最佳反应时间。

表5 反应时间对收率的影响
2.2.4丙炔醇用量对收率的影响
在100 mL三口烧瓶内加入2.66 g苄基叠氮、0.352 g抗坏血酸、0.10 g CuSO4·5H2O,加入5 mL蒸馏水和20 mL四氢呋喃,改变丙炔醇用量,63℃下恒温反应24 h。实验结果如表6所示,增加丙炔醇用量[n(丙炔醇)∶n(叠氢钠)增加],产物收率基本呈直线上升,在比值为1∶1.2时有最大收率为87.79%;继续增加丙炔醇,产物收率变化不明显,由此可确定最佳n(丙炔醇)∶n(叠氮钠)为1∶1.2。

表6 丙炔醇用量对收率的影响
2.31-苄基-4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1,2,3-三氮唑(Ⅲ)的合成
2.3.1反应温度对收率的影响
在100 mL三口烧瓶中,依次加入γ-氯丙基三甲氧基硅烷1.985 g、金属钠0.345 g、1-苄基-3-羟甲基-1H-1,2,3,-三氮唑(Ⅱ)1.89 g、二氯甲烷30 mL,改变反应温度,磁力搅拌下反应8 h。由表7可知,收率随反应温度升高而升高,到40℃达到最大值,继续升高温度,受二氯甲烷沸点限制,收率基本不变,故40℃为最佳反应温度。

表7 反应温度对收率的影响
2.3.2二氯甲烷用量对收率的影响
在100 mL三口烧瓶中,依次加入γ-氯丙基三甲氧基硅烷1.985 g、金属钠0.345 g、1-苄基-3-羟甲基-1H-1,2,3,-三氮唑(Ⅱ)1.89 g,改变二氯甲烷用量,40℃下反应8 h。由表8可知,随着溶剂用量增多,反应物浓度下降,导致收率下降,故20 mL为最佳溶剂用量。

表8 二氯甲烷用量对收率的影响
2.3.3反应时间对收率的影响
在100 mL三口烧瓶中,依次加入γ-氯丙基三甲氧基硅烷1.985 g、金属钠0.345 g、1-苄基-3-羟甲基-1H-1,2,3,-三氮唑(Ⅱ)1.89 g、二氯甲烷20 mL,反应温度设定为40℃,改变反应时间进行实验。由表9可知,收率随反应时间增长而变大,到12 h达到最大值,继续增长反应时间,由于产生副产物造成收率下降,所以12 h为最佳反应时间。

表9 反应时间对收率的影响
2.44-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑(Ⅳ)的合成
2.4.1反应温度对收率的影响
在100 mL三口烧瓶中,加入1-苄基-3-羟甲基-1H-1,2,3,-三氮唑(Ⅲ)3.51 g、三氟乙酸30 mL,改变反应温度,磁力搅拌下反应36 h。由表10可知,随温度升高收率不断增大,达到70℃后变化不大,查阅资料得知三氟乙酸沸点约为72℃,继续升高温度溶液温度将不再升高,所以72℃为最佳反应温度。

表10 反应温度对收率的影响
2.4.2三氟乙酸用量对收率的影响
在100 mL三口烧瓶中加入1-苄基-3-羟甲基-1H-1,2,3,-三氮唑(Ⅲ)3.51 g,改变三氟乙酸用量,在72℃恒温下反应36 h。由表11可知,三氟乙酸用量低于20 mL时,收率随三氟乙酸用量增加而变大,这是因为溶剂用量过低,反应物料不足以充分接触导致收率较低;继续增加三氟乙酸用量后,反应物料浓度均降低,收率反而下降。可以确定20 mL为最佳三氟乙酸用量。

表11 三氟乙酸用量对收率的影响
2.4.3反应时间对收率的影响
100 mL三口烧瓶中依次加入1-苄基-4-{[3-(三甲氧基硅烷基)1H-1,2,3-]甲基}-1,2,3-三氮唑(Ⅲ)3.51 g、三氟乙酸20 mL,设定反应温度72℃,在不同的反应时间下进行实验。由表12可知,随反应时间增加,反应越来越充分,收率不断增加,到36 h时达到最大值,继续反应会发生副反应致使收率降低。故选择36 h为最佳反应时间。

表12 反应时间对收率的影响
3 结论
本文以氯化苄为原料,通过叠氮取代和1,3-偶极环加成反应得到N-保护剂-1,2,3-三氮唑然后接枝在γ-氯丙基三甲氧基硅烷上,再脱除保护剂,最后获得了可用于制备质子交换膜的大单体4-{[3-(三甲氧基硅烷基)丙氧基]甲基}-1H-1,2,3-三氮唑。本合成路线原料廉价易得、反应条件温和、环境污染少且后处理简单。
参考文献:
[1]石建恒,于宏燕,曾心苗.燃料电池质子交换膜的研究现状[J].膜科学与现状,2009,29(2):94-98.
[2]Shao Zhigang,Joghee Prabhuram,Hsing I-Ming.Preparation and characterization of hybrid Nafion-silica membrane doped with phosphotungstic acid for high temperature operation of proton exchange membrane fuel cells [J].Journal of Membrane Science,2004,229:43-51.
[3]Peighambardoust S J,Rowshanzamir S,Amjadi M.Review of the proton exchange membranes for for fuel cell applications[J].International Jouranl of Hydrogen Energy,201,35:9349-9348.
[4]Matos B R,Arico'AE E M,Linardi AE M.Thermal properties of Nafion-TiO2composite electrolytes for PEM fuel cell[J].J Therm Anal Calorim,2009,97:591-594.
[5]Maalouf M,Ghassemi H,Lee C G,et al.Triazole-based electrolytes for fuel cell applications:material properties and proton transfer capabilities[J].J ECS Transactions,2009,51(6):383-388.
[6]Subbaraman R,Ghassemi H,Zawodzinski Jr T.Triazole and triazole derivatives as proton transport facilitators in polymer electrolyte membrane fuel cells[J].J Solid State Ionics,2009,180:1143-1150.
[7]Zhou Zhen,Li Siwen,Zhang Yuelan,et al.Promotion of proton conduction in polymer electrolyte membranes by 1H-1,2,3-Triazole[J].J Am Chem Soc,2005,127 (31):10824-10825.
[8]Shilpi Sanghi,Mark Tuominen,Bryan Coughlin E.Hybrid inorganic-organic proton exchange membranes containing 1H-1,2,3-Triazole moieties[J].J Solid State I-onics,2010,181:1183-1188.
[9]Loren J C,Krasinski A,Fokin V V,et al.NH-1,2,3-triazoles from azidomethyl pivalate and carbamates:Base -Labile.N-Protecting groups[J].J Synlett,2005,18:2847-2850.
中图分类号:TQ21
文献标识码:A
文章编号:1003-3467(2016)04-0021-05
收稿日期:2016-02-11
基金项目:河南省国际合作项目(104300510009)
作者简介:毛桢东(1989-),男,在读硕士,研究方向为新型能源材料的开发与应用,电话:13598868012。
Synthesis and Characterization of 4-{[3-(Trimethoxysilyl)Propoxy]Methyl}-1H-1,2,3-Triazole
MAO Zhendong,WANG Lei,GAO Pengjie,YU Xiuli,LI Huiping
(School of Chemical Engineering and Energy,Zhengzhou University,Zhengzhou450001,China)
Abstract:Using benzyl chloride as raw materials,through the azide replaced generate benzyl azide(Ⅰ),then click on the chemical synthesis of 1-benzyl-3-hydroxy methyl-1H-1,2,3-triazole(Ⅱ),secondly using Williamson reaction to gamma chloride propyl silane with three oxygen radicals generated(Ⅱ)grafted 1-benzyl-4-{[3-(trimethoxysilyl)propoxy]methyl}-1,2,3-triazole(Ⅲ),the final removal benzyl get 4-{[3-(trimethoxysilyl)propoxy]methyl}-1H-1,2,3-triazole(Ⅳ),it can be used in the preparation of fuel cell proton exchange membrane.The structure of product is characterized by FT-IR,NMR,the results confirm the feasibility of synthetic route.Using single factor experiment and orthogonal experiment to investigate the reaction temperature,reaction time,reaction material mole ratio and dosage of catalyst,the optimum technological conditions for the average yield of each step are 83.89%,92.60%,90.56%and 71.39%respectively.
Key words:click chemistry;4-{[3-(trimethoxysilyl)propoxy]methyl}-1H-1,2,3-triazole;PEM
猜你喜欢
化学工程师(2022年3期)2022-04-19
上海化工(2021年2期)2021-04-23
商品与质量(2019年32期)2019-11-29
火工品(2018年1期)2018-05-03
中国资源综合利用(2017年3期)2018-01-22
科技与企业(2015年20期)2015-10-21
中国洗涤用品工业(2015年9期)2015-02-28
中国塑料(2014年10期)2014-10-17
天然产物研究与开发(2014年6期)2014-04-27
火炸药学报(2014年3期)2014-03-20
