
离子色谱法测定核电站一回路冷却剂中痕量F-,Cl-和
2016-08-19 04:00韩剑三门核电有限公司浙江台州317112
化学分析计量 2016年4期
韩剑(三门核电有限公司,浙江台州 317112)
离子色谱法测定核电站一回路冷却剂中痕量F-,Cl-和
韩剑
(三门核电有限公司,浙江台州 317112)
建立离子色谱法测定核电站一回路冷却剂中痕量氟离子F-,氯离子Cl-,硫酸根离子的方法。采用80 mmol/L硼酸溶液配合氢氧化钾淋洗液发生器在线生成淋洗液,梯度洗脱,淋洗液流量为1.2 mL/min,在选定的分析条件下,和不干扰F-,Cl-,的测定。F-,Cl-,的质量浓度与其色谱峰面积呈良好的线性关系,线性相关系数分别为0.999 8,0.999 5,0.999 7,线性范围分别为0.85~30.0,2.65~30.0,2.00~30.0 μg/L。F-,Cl-,测定结果的相对标准偏差分别为0.56%~1.58%,0.85%~3.62%,1.21%~4.60%(n=7)。F-, Cl-,的加标回收率分别为98%~104%,98%~108%,93%~108%。该方法快速、准确,满足核电站一回路冷却剂中痕量F-,Cl-,的检测要求。
氟离子;氯离子;硫酸根离子;硼酸;冷却剂;离子色谱法
如果压水堆核电站一回路冷却剂中含有大量的F-,Cl-,等阴离子或其它有害杂质,会使一回路燃料包壳的锆合金及其它设备和管道的不锈钢、镍基合金、低合金钢、碳钢等结构材料受到腐蚀损坏,一方面影响反应堆的正常运行,可能造成非计划停堆,导致巨大的经济损失;另一方面可能造成反应堆中放射性物质的泄漏,对人员和环境造成危害[1-2]。因此准确检测一回路冷却剂中痕量F-,Cl-,等阴离子非常重要。
离子色谱法分析阴离子和阳离子具有分析快速,灵敏度高,选择性好和同时测定多组分的优点[3-5]。但压水堆核电站在运行过程中,一回路冷却剂中需添加硼酸和氢氧化锂,调节反应和抑制系统材料的腐蚀,并且两者的浓度随机组运行不断变化[6]。如果采用单一的氢氧化钾淋洗液体系分析一回路冷却剂样品,由于氢氧化钾淋洗液与样品中的硼酸发生化学反应生成四硼酸钾,使淋洗液的组分发生变化,导致样品中F-,Cl-,的色谱峰受到干扰并且保留时间发生漂移,无法对其进行定性和定量分析。检测一回路冷却剂中痕量F-,Cl-,的常见方法是使用四硼酸钠作为淋洗液进行梯度淋洗,通过四硼酸钠和纯水比例的变化改变淋洗过程中四硼酸钠的浓度,进而实现对F-,Cl-,的分离和定量分析,但该法具有一定的局限性,配制四硼酸钠淋洗液时因人工操作不同而使其浓度存在误差,淋洗液也容易吸收二氧化碳而产生碳酸根污染,导致待测离子的保留时间不稳定,重复性差,使检测结果不稳定[7]。当用四硼酸钠淋洗液梯度淋洗时,抑制产物硼酸的浓度会随着梯度的变化而升高,从而造成基线漂移,而使检测结果不稳定[8]。笔者建立了以80 mmol/L硼酸溶液配合氢氧化钾淋洗液发生器在线生成淋洗液的方式,在保持硼酸淋洗液浓度不变的情况下改变氢氧化钾浓度,硼酸和氢氧化钾发生化学反应生成不同浓度的四硼酸钾淋洗液。与使用四硼酸钠作为淋洗液的分析方法比较,既避免了碳酸根对淋洗液的污染,也降低了人工配制淋洗液产生的误差对检测结果稳定性的影响,不仅可以得到稳定的背景电导,将基线漂移降到最小,而且可以得到稳定的峰面积响应值,使色谱峰的积分更准确,从而保证了分析数据的可靠性。
1 实验部分
1.1主要仪器与试剂
超纯水仪:Advantage A10型,美国密理博公司;
离子色谱仪:ICS-5000型,配置AXP进样泵,自带脱气腔的四元梯度分析泵,带有氢氧化钾淋洗液在线发生器储罐的EG-KOH淋洗液发生装置、阴离子捕获柱、阴离子保护柱、阴离子分离柱、阴离子抑制器AERS 500、二氧化碳去除装置CRD 200(4 mm)、电导检测器、绿色PEEK管制作的1 mL定量环,美国戴安公司;
硼酸:纯度为99.999 9%,德国默克公司;
氢氧化锂(一水):纯度为99.995%,美国阿拉丁公司;
F-标准溶液:1 000 mg/L,相对扩展不确定度为1%(k=2),中国计量科学研究院;
Cl-标准溶液:1 000 mg/L,相对扩展不确定度为0.7%(k=2),中国计量科学研究院;标准溶液:1 000 mg/L,相对扩展不确定度为0.7%(k=2),中国计量科学研究院;
硼酸溶液:80 mmol/L,准确称取4.946 4 g高纯硼酸,用超纯水溶解后转移到1 000 mL容量瓶中,定容至刻标线;
氢氧化锂溶液(用于模拟一回路样品):100 mg/L,准确称取0.604 6 g氢氧化锂(一水),用超纯水溶解后转移到1 000 mL容量瓶中,定容至标线;
硼基体溶液(用于模拟一回路样品):10 000 mg/L,准确称取57.091 4 g高纯硼酸,用超纯水溶解后转移到1 000 mL容量瓶中,定容至标线;
实验用水为超纯水。
1.2仪器工作条件
色谱柱:阴离子捕获柱为ATC-3(24 mm×9 mm,美国戴安公司),阴离子保护柱为IonPac AG14 Guard Column(50 mm×4 mm,美国戴安公司),阴离子分离柱为IonPac AS14 Analytical Column(250 mm×4 mm,美国戴安公司);淋洗液:由80 mmol/L硼酸溶液和不同浓度的氢氧化钾溶液(浓度见表1)在EG-KOH淋洗液发生装置反应生成不同浓度的四硼酸钾淋洗液,梯度洗脱,淋洗液流量为1.2 mL/min,淋洗液氮气加压35~70 kPa(8~10 psi);柱温箱温度:25℃;色谱柱温度:30℃;电导池温度:35℃;抑制器:抑制电流为120 mA,外加水模式活化,氮气加压35~70 kPa(8~10 psi);进样方式:1 mL定量环。
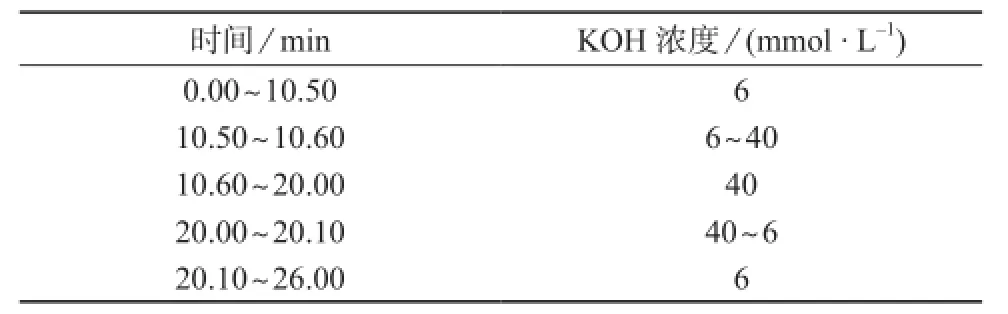
表1 梯度淋洗的KOH浓度
1.3标准工作溶液的配制
分别移取1 mL F-,Cl-,标准溶液,用超纯水定容至1 000 mL,配制成1 mg/L的F-,Cl-,标准工作储备液。用超纯水将1 mg/L的F-, Cl-,标准工作储备液稀释成浓度均为5,10, 15,20,25,30 μg/L的系列混合标准溶液。
2 结果与讨论
2.1仪器工作条件的选择
在离子色谱分析中,淋洗液流速和浓度、分离系统温度均会对待测离子的保留时间和分离度产生影响[9-10],结合待测离子和干扰离子的保留特性及特点,经过调试选择淋洗液流速为1.2 mL/min,选择80 mmol/L硼酸溶液与表1中梯度变化的氢氧化钾浓度在线生成梯度变化的四硼酸钾淋洗液,选择柱箱温度和色谱柱温度分别为25℃和30℃。待测离子的定量是基于其峰面积的大小,在选定的淋洗液流速和浓度条件下,进样量和电导池温度均会对色谱峰的峰面积产生影响,经调试选择进样量为1 mL,电导池温度为35℃。
根据硼酸和氢氧化钾发生化学反应的比例关系,4 mmol/L硼酸与2 mmol/L氢氧化钾生成1 mmol/L的四硼酸钾,在整个分析过程中四硼酸钾的浓度是依据氢氧化钾的变化而变化的。当氢氧化钾浓度为6 mmol/L时淋洗液组分为3 mmol/L的四硼酸钾和过剩的硼酸溶液;当氢氧化钾为40 mmol/L时淋洗液组分为20 mmol/L的四硼酸钾溶液,硼酸溶液全部参与反应,没有过剩的硼酸溶液;当氢氧化钾浓度在6~40 mmol/L之间梯度变化时,四硼酸钾和过剩硼酸的浓度也在梯度变化。结合淋洗液的浓度及梯度变化过程,选择抑制器电流为120 mA,抑制器采用外加水模式,以纯水作为阴极侧和阳极侧的电解液,而不是将经电导池测量后流出的废液作为抑制器的电解液。
在选定的实验条件下,仪器的背景电导稳定在3.235~3.658 μS之间,系统压力稳定在14.4~14.7 MPa (2 094~2 128 psi)之间。
2.2方法的干扰及消除
在待测的F-,Cl-,标准溶液中加入干扰离子,进样分析,色谱图如图1所示。在选定的仪器配置及实验条件下,该方法对F-, Cl-,和有很好的分离和选择性。虽然进样体积较大(1 mL),导致出现一个较大的水负峰,但在对F-进行积分时并不受水负峰干扰,并且与弱保留的有机酸离子可以很好地分离。虽然和的色谱峰重合,但由于和不是检测目标,不干扰F-,Cl-,的测量,因此不予考虑。
在核电站正常运行过程中一回路冷却剂中添加了硼酸和氢氧化锂,并且硼酸浓度和氢氧化锂浓度不断变化[6]。为验证加入硼酸和氢氧化锂是否对待测离子积分有影响,分别配制10 μg/L F-,Cl-,标准溶液,硼含量为1 000 mg/L的10 μg/L F-,Cl-,标准溶液,硼含量为1 700 mg/L和氢氧化锂含量为4 mg/L(以Li计)的10 μg/L F-, Cl-,标准溶液,进样分析后的色谱图如图2~图4所示。从图2~图4可见,随着硼浓度的增加,水负峰区域的基线电导产生了明显的变化,从一个较大的负峰逐渐变化为一个较大的正峰,但其并未对待测离子的积分产生影响。从图3、图4可见,氢氧化锂的加入未对待测离子的积分产生影响。因此硼酸和氢氧化锂的加入未对待测离子的积分产生影响。

图4 添加B和LiOH后F-,Cl-标准溶液色谱图
2.3线性方程
将1.3中标准工作溶液分别进样分析获得不同质量浓度与其相应的峰面积响。以各离子的浓度(μg/L)为横坐标、峰面积(μS·min)为纵坐标绘制工作曲线,拟合时选择不包含原点线性拟合,各离子的线性关系良好,线性方程、线性范围和线性相关系数见表2。
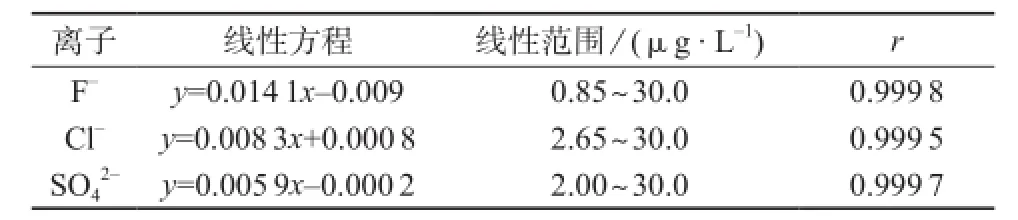
表2 线性方程、线性范围和相关系数
2.4方法检出限和定量下限
按照HJ 168-2010要求[11-13],进行9次空白样品的分析,由于空白样品中只检测出F-,因此F-的方法检出限(MDL)和定量下限(LLD)以空白样品的测定结果计算。按照预估方法检出限的3~10倍配制9个加标平行样,以加标平行样Cl-,SO42-的测定结果计算各自的MDL和LLD。计算时采用99%(双侧)置信度,按照5倍的MDL计算LLD,各离子的测定结果及相关数据见表3。
表3 F-,Cl-,的检出限和定量下限
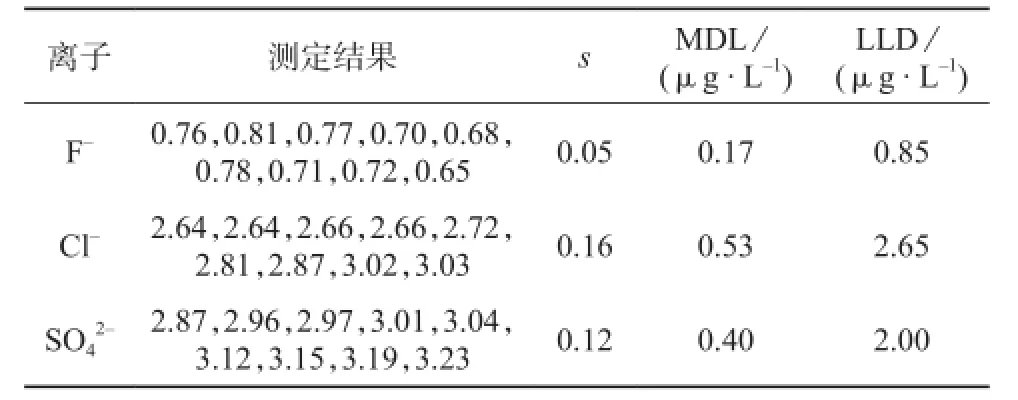
表3 F-,Cl-,的检出限和定量下限
离子 测定结果 s MDL/ (μg·L-1)LLD/ (μg·L-1)F- 0.76,0.81,0.77,0.70,0.68,0.78,0.71,0.72,0.65 0.05 0.17 0.85 Cl- 2.64,2.64,2.66,2.66,2.72,2.81,2.87,3.02,3.03 0.16 0.53 2.65 SO42- 2.87,2.96,2.97,3.01,3.04,3.12,3.15,3.19,3.23 0.12 0.40 2.00
2.5一回路冷却剂的模拟样品分析
为了确定硼锂浓度的不断变化是否会对阴离子痕量分析的准确度产生影响,根据三门核电AP1000机组运行中一回路水化学控制的特点,按照300℃下硼锂协调曲线中的硼锂浓度的变化关系配制一回路冷却剂的模拟水样,分别配制10 μg/L F-,Cl-,标准溶液;加入硼酸和氢氧化锂的10 μg/L F-,Cl-,标准溶液;加入硼酸和氢氧化锂的20 μg/L F-,Cl-,标准溶液。用该实验方法对各个样品进行分析,将10 μg/L F-,Cl-,标准溶液的分析结果作为加标量,另外两个标准溶液的分析结果的差值作为回收值,F-,Cl-,的分析结果及回收率分别见表4~表6。
从表4~表6中的数据可知,F-的加标回收率为99%~105%,Cl-的加标回收率为95%~104%,的加标回收率为95%~107%,3种离子的加标回收率均在90%~110%之间,说明硼、锂浓度的不断变化对F-,Cl-,的分析准确度未产生影响。

表4 硼锂浓度变化时F-测定结果
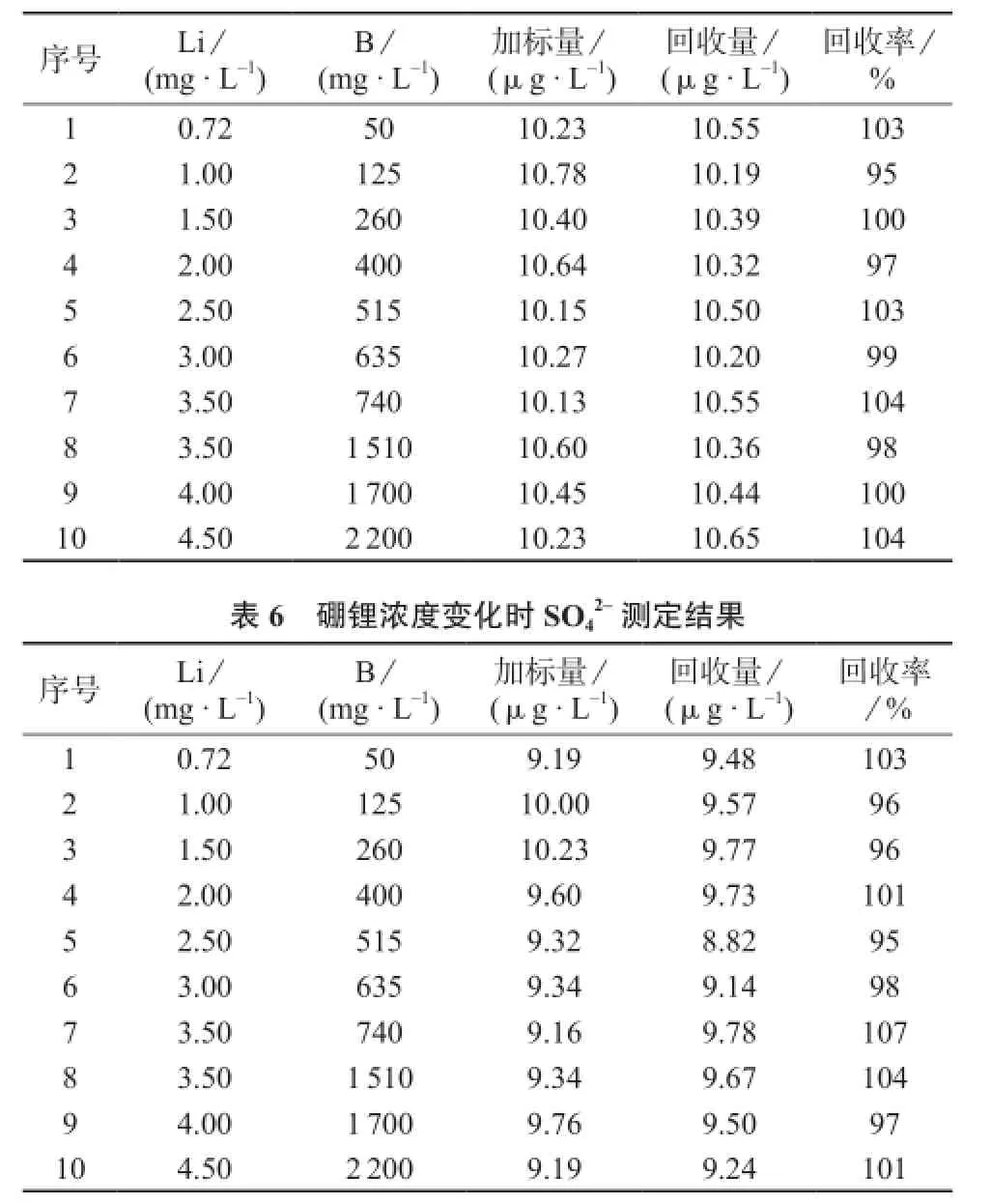
表5 硼锂浓度变化时Cl-测定结果
2.6方法准确度和精密度
以10 μg/L F-,Cl-,标准溶液为基体,分别配制7个F-,Cl-加标量为5,10,15 μg/L的平行样,每个平行样的测定结果减去10 μg/L F-, Cl-,标准溶液的测定结果为加标回收量计算加标回收率,测定及计算结果见表7。
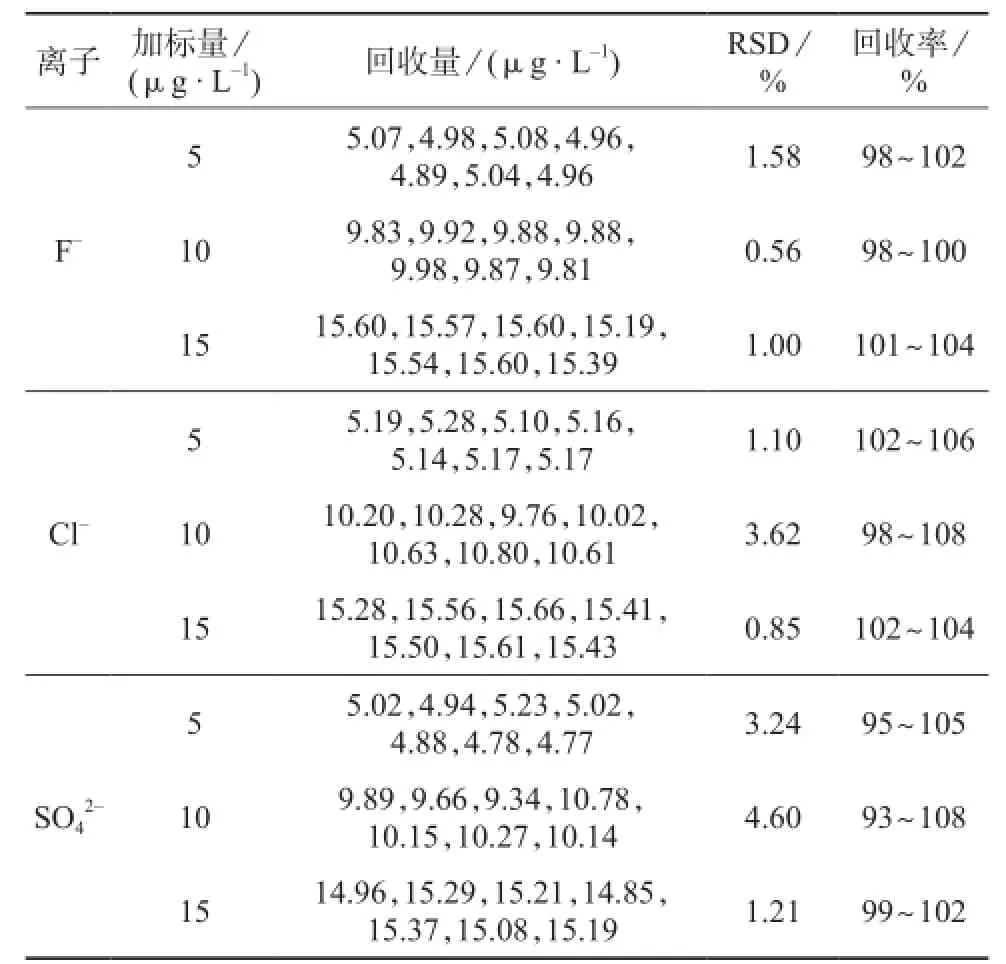
表7 方法精密度和回收试验结果
由表7中的试验数据可以看出,F-的加标回收率为98%~104%,测定结果的相对标准偏差为0.56%~1.58%;Cl-的加标回收率为98%~108%,测定结果的相对标准偏差为0.85%~3.62%;的加标回收率为93%~108%,测定结果的相对标准偏差为1.21%~4.60%。F-,Cl-,的加标回收率都在90%~110%之间,测定结果的相对标准偏差均小于5%,表明该方法具有较高的准确度和精密度,满足分析方法的要求。
3 结语
采用80 mmol/L硼酸溶液配合氢氧化钾淋洗液发生器在线生成淋洗液的方式,建立了分析三门核电AP1000机组一回路冷却剂中痕量F-,Cl-,SO42-的方法。通过优化仪器配置和条件参数,在选定的实验条件下,该方法的线性系数、检出限、定量范围、精密度及回收率均能满足分析检测的要求,能快速、准确地测定AP1000机组一回路冷却剂中痕量F-,Cl-,
[1] 白新德.核材料化学[M].北京:化学工业出版社,2007: 391-466.
[2] 韩延德.核电厂水化学[M].北京:原子能出版社,2010: 30-42.
[3] 牟世芬,刘克纳,丁晓静.离子色谱方法及应用[M].北京:化学工业出版社,2005: 1-5.
[4] 鲁蕴甜.离子色谱在环境分析中应用概述[J].广东化工,2014,41(11): 166-167.
[5] 潘灿盛,李维嘉,吴凌涛,等.离子色谱在饮用水及食品分析中的应用研究[J].广州化工,2014,42(7): 33-34,95.
[6] 宋利君,刘飞华,李成涛,等. B-Li水化学对核电站金属材料腐蚀的影响[J].核科学与工程,2014,34(1): 97-101,115.
[7] 蔡艳艳,于军波,马艳,等.离子色谱技术的发展及其在食品添加剂检测中的应用[J].分析仪器,2013(6): 1-6.
[8] 李昌厚.高效液相色谱研发和应用中关键问题的研究[J].分析仪器,2013(3): 44-49.
[9] 张燕,韩秉均,唐新建,等.火电机组水汽中痕量阴离子的毛细管离子色谱法测定[J].热力发电,2015(9): 19-25.
[10] 余秀娟,曾钰,那晶晶,等.离子色谱法同时测定水中8种阴离子[J].环保科技,2015(4): 57-59.
[11] 蒋晓光,王艳君,王彩云,等.硫代硫酸钠滴定法连续测定铜磁铁矿中铜和铁[J].化学分析计量,2015,24(3): 22-26.
[12] 曾兴宇,刘静,周东星.紫外消解流动注射光度法测定海水养殖废水中总氮、总磷[J].化学分析计量,2015,24(3): 62-65.
[13] HJ 168-2010 环境监测分析方法标准制修订技术导则[S].
Determination of Trace F-, Cl-,in Power Plant Primary Coolant by Ion Chromatography
Han Jian
(Sanmen Nuclear Power Co., Ltd., Taizhou 317112, China)
The method was established for determination of trace F-,Cl-and-in power plant primary coolant by ion chromatography. The 80 mmol/L boric acid was coordinated with the KOH eluent generator to produce new eluent online for gradient elution,and eluent flow rate was 1.2 mL/min. Under the selected conditions,the determination of F-, Cl-andwere not influenced byand. The mass concentration of F-,Cl-andwere linear with its peak areas. The correlation coefficients of F-,Cl-,were 0.999 8, 0.999 5, 0.999 7, and the detection limits were 0.85-30.0,2.65-30.0,2.00-30.0 μg/L, respectively. The relative standard deviations of F-,Cl-,detection results were 0.56%-1.58%,0.85%-3.62%, 1.21%-4.60%(n=7), respectively. The recoveries of F-,Cl-andwere 98%-104%, 98%-108%, 93%-108%, respectively. The method can be used for detecting trace F-,Cl-,in power plant primary coolant quickly and accurately.
F-; Cl-; boric acid; coolant; ion chromatography
O657.7
A
1008-6145(2016)04-0064-04
10.3969/j.issn.1008-6145.2016.04.017
联系人:韩剑;E-mail: hanj2@smnpc.com.cn
2016-05-30
猜你喜欢
水泵技术(2022年3期)2022-08-26
盐科学与化工(2022年5期)2022-05-19
世界有色金属(2021年6期)2021-06-14
中国金属通报(2020年9期)2020-12-30
现代食品·下(2019年12期)2019-10-21
中国核电(2017年1期)2017-05-17
中国核电(2017年1期)2017-05-17
化学教学(2017年3期)2017-04-21
汽车文摘(2016年5期)2016-05-31
分析化学(2015年10期)2015-11-03
