
核磁共振法测定片剂中西咪替丁*
2016-08-19 03:59:55张秀丽王聪王远红吕志华中国海洋大学医药学院山东青岛266003
化学分析计量 2016年4期
张秀丽,王聪,王远红,吕志华(中国海洋大学医药学院,山东青岛 266003)
核磁共振法测定片剂中西咪替丁*
张秀丽,王聪,王远红,吕志华
(中国海洋大学医药学院,山东青岛 266003)
建立以核磁共振技术测定片剂中西咪替丁含量的方法。采用Agilent DD2-500型核磁共振波谱仪,以氘代甲醇为溶剂、对苯二甲酸二甲酯为内标,测试温度25℃,弛豫时间为20 s,脉冲角为45°,采集时间为2 s,扫描次数为16次,采集核磁共振氢谱。该方法线性范围为0.1~5.0 mg/mL,相关系数r=0.999 8,测定结果的相对标准偏差为0.11%(n=6),平均加标回收率在100.03%~100.58%之间。用该方法测定不同厂家片剂中西咪替丁的含量,测定结果与药典方法相吻合。该方法简单快速、样品用量少,适用于西咪替丁的质量控制。
西咪替丁;核磁共振法;片剂;定量分析
西咪替丁(Cimetidine)又名甲氰咪胍(化学结构式见图1),是一种H2受体阻滞剂,可抑制胃酸分泌,临床上主要用于胃酸过多、消化道溃疡的治疗。近年来西咪替丁用于非溃疡性疾病的治疗又有新发现,如对慢性乙型肝炎的治疗,对烧伤引起的免疫作用[1-2]等。目前测定西咪替丁的方法有高效液相色谱法[3-4]、分光光度法[5]、HPLC-UV[6]、色谱-质谱法[7]等,美国药典USP35版中采用高效液相色谱法[8],中国药典2015年版中采用分光光度法[9]。高效液相色谱法一般以乙腈为流动相,由于西咪替丁带有-NH-等极性基团,比较容易产生拖尾,为了改良峰型,需要加入三乙胺或磷酸盐为消尾剂,这样会损害色谱柱减少色谱柱的使用寿命。分光光度法样品配制步骤繁琐,且需消耗大量试剂,准确度相对不高。2010年《中华人民共和国药典》中新增了核磁共振定量法,核磁共振定量技术具有准确、简单、快速,无需分离提取,不破坏被测样品等优势[10-16],除能进行定量分析外,还能同时通过结构分析鉴别假冒伪劣药品。目前核磁共振技术测定西咪替丁含量的方法还未见文献报道。笔者建立了核磁共振定量技术测定片剂中西咪替丁含量的方法,考察并优化了实验条件。利用该方法测定了不同厂家片剂中西咪替丁的含量,与文献方法相比,更为快速、简单,且无需对照品。所建方法可为西咪替丁的质量控制提供新技术和新方法。

图1 西咪替丁的化学结构式
1 实验部分
1.1主要仪器与试剂
核磁共振波谱仪:Agilent 500-DD2型,美国安捷伦科技有限公司;
电子天平:BT25S型,德国赛多利斯科学仪器北京有限公司;
紫外可见分光光度仪:2802PCS型,美国尤尼柯上海仪器有限公司;
氘代甲醇:氘代度大于99.8%,美国Sigma-Aldrich公司;
西咪替丁片剂:规格0.2 g,批号为141102,山东仁和堂药业有限公司;
西咪替丁片剂:规格0.2 g,批号为1509262,河南中杰药业有限公司;
西咪替丁对照品:纯度为99%,日本东京化成工业株式会社;
对苯二甲酸二甲酯:纯度99.99%,西格玛奥德里奇上海贸易有限公司。
1.2核磁共振氢谱定量实验参数
温度:25℃;弛豫时间:20 s;脉冲角:45°;采集时间:2 s;扫描次数:16次;谱宽:8 012.8 Hz。
1.3核磁共振定量方法
1.3.1标准溶液及试样溶液配制
内标溶液的配制:精密称取对苯二甲酸二甲酯6.25 mg,置于25 mL容量瓶中,用氘代甲醇定容,振荡混匀,配制成0.25 mg/mL的内标溶液。
西咪替丁对照品储备液:精密称取西咪替丁对照品5.00 mg,加入1.0 mL上述内标溶液溶解,配制成5 mg/mL的西咪替丁对照品储备液,以此配制系列浓度的标准溶液。
西咪替丁供试品溶液的配制:取西咪替丁20片,精密称量,研细,精密称取样品粉末3.00 mg,加入1.0 mL上述内标溶液,振荡混匀后,静置5 min,待辅料沉淀于底部,取0.5 mL上清液于5 mm核磁管中,进行定量1H NMR测试。
1.3.2结果计算
西咪替丁的量按式(1)计算:

式中:Wu——西咪替丁的质量,mg;
Au——西咪替丁的核磁共振峰面积;
As——内标物质的核磁共振峰面积;
Ws——内标物的质量,mg; Eu——西咪替丁的质子当量(样品的相对分子质量与样品定量峰相应基团的质子数之比);
Es——内标物的质子当量(内标的相对分子质量与内标定量峰相应基团的质子数之比)。
1.4紫外分光光度法定量方法
按照2015年版中国药典方法,取西咪替丁20片,精密称量,研细。精密称取50 mg置于50 mL容量瓶中,用0.01 mol/L的盐酸溶液定容摇匀,滤过,精密移取0.5 mL续滤液,置于50 mL容量瓶中,再用上述溶剂稀释至标线,摇匀。采用紫外分光光度法,在218 nm波长处测定吸光度,按C10H16N6S的吸收系数为774计算,即得西咪替丁含量。
2 结果与讨论
2.1溶剂的选择
西咪替丁和对苯二甲酸二甲酯(内标物)在氘代试剂中溶解性均良好,可防止样品部分溶解带来的误差,而且片剂中辅料在氘代甲醇中均不溶解,可以避免辅料共振峰对定量的干扰,因此实验选择以氘代甲醇作为溶剂。
2.2内标物的选择
内标物应具有较高的纯度及较少的质子数,主要吸收峰与样品峰之间无干扰,且不与样品反应[11]。根据西咪替丁的化学结构与核磁共振氢谱,实验选择以对苯二甲酸二甲酯作为内标。对苯二甲酸二甲酯在氘代甲醇中很稳定且混合后不反应,该化合物的核磁共振氢谱只有两组共振峰,并且与西咪替丁样品的共振峰完全分离,互不干扰(见图2),满足定量要求。
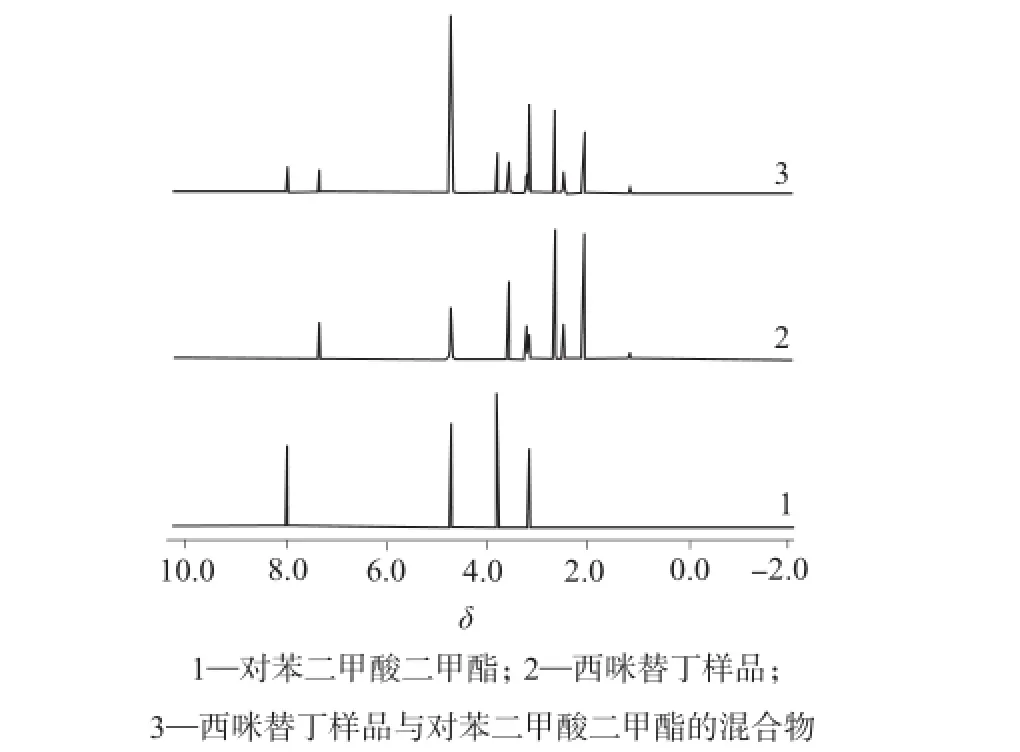
图2 西咪替丁样品、内标及二者混合后的核磁共振氢谱
2.3定量峰的选择
通常选择与相邻峰无干扰且裂分较少的峰作为定量峰,且尽量避免选择甲基峰,减少定量误差。根据二者的化学结构,本实验选择布西咪替丁咪唑环上δ7.47处共振峰和对苯二甲酸二甲酯苯环δ8.10处共振峰为定量峰,二者化学环境相似,共振峰均在低场区,且完全分离(见图3),满足定量要求。
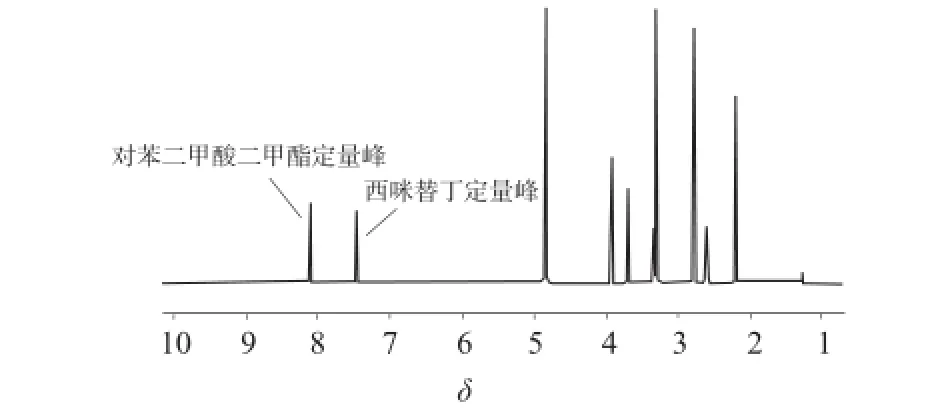
图3 西咪替丁与对苯二甲酸二甲酯混合溶液的核磁共振氢谱
2.4仪器参数的优化
2.4.1弛豫时间
试验考察了弛豫时间(d1)为4,10,15,20,25,30,40 s时对核磁定量的影响。结果发现,d1≤15 s时,随着d1的增加,样品和内标物的定量峰积分面积比值逐渐增大;当d1>15 s时,样品和内标物的定量峰积分面积比值没有明显变化,因此实验选择d1为20 s。
2.4.2扫描次数
分别设置扫描次数(nt)为4,8,16,32,64,128次进行试验,结果表明,随着扫描次数的改变,样品和内标物积分面积的比值没有明显变化,扫描次数越多测试时间越长。在满足信噪比的条件下,实验选择扫描次数为16次。
2.4.3采集时间
采集时间at与谱图的数字分辨率有很大的关系。合适的采集时间不仅可以保证FID信号衰减完全,同时也能够提高仪器的使用效率,采集时间一般选择范围为2~4 s[17]。试验考察了at分别为2,3,4 s时对核磁共振氢谱的影响,结果表明,当at=2 s 时FID 信号衰减完全,满足定量要求。因此实验选择采集时间为2 s。
2.5精密度试验
配制1份西咪替丁供试品溶液,重复测定6次,对西咪替丁δ7.47处共振峰和对苯二甲酸二甲酯苯环δ8.10处共振峰进行积分,以二者的面积比计算检测精密度,试验结果见表1。由表1可知,6次测定结果的相对标准偏差为0.11%,说明该方法精密度良好。

表1 精密度、重复性及稳定性试验结果
2.6重复性试验
精确称取6份样品,用0.25 mg/mL的内标液溶解,取上清液测定核磁共振氢谱,以西咪替丁和内标物定量峰面积比值计算西咪替丁含量并检测重复性,试验结果列于表1。由表1可知,6次测定结果的相对标准偏差为0.33%,说明所建方法的重复性良好。
2.7稳定性试验
将2.5中西咪替丁供试品溶液,分别放置0,4,8,24,48,72 h后进行测定,计算样品和内标定量峰面积比值,结果列于表1。由表1可知,6次测定结果的相对标准偏差为0.45%,表明该样品在室温放置72 h内稳定,满足分析测试要求。
2.8曲线方程
用内标溶液对西咪替丁对照品储备液进行稀释,配制成质量浓度分别为5,2.5,1.0,0.5,0.25,0.1 mg/mL的西咪替丁系列标准溶液,分别测定。以西咪替丁与内标定量峰面积的比值(y)为纵坐标,以西咪替丁质量浓度(x)为横坐标进行线性回归,得回归方程y=0.370 7x-0.009 7,相关系数r=0.999 8。当内标物的质量浓度为0.25 mg/mL时,西咪替丁在0.1~5.0 mg/mL的范围内线性良好。
2.9检出限与定量限
根据核磁共振氢谱,以信噪比S/N=3时的测定浓度为检出限(LOD);以信噪比S/N=10时的测定浓度为定量限(LOQ)。该方法的LOD为10 µg/mL,LOQ为28.57 µg/mL。
2.10回收试验
分别在西咪替丁供试品溶液中加入高、中、低浓度的西咪替丁标准溶液,每种浓度配制3份平行样,进行核磁共振氢谱测试并计算回收率,结果见表2。由表2可知,3个加标水平下,平均回收率分别为100.51%,100.03%,100.58%,说明该方法具有较高准确度。
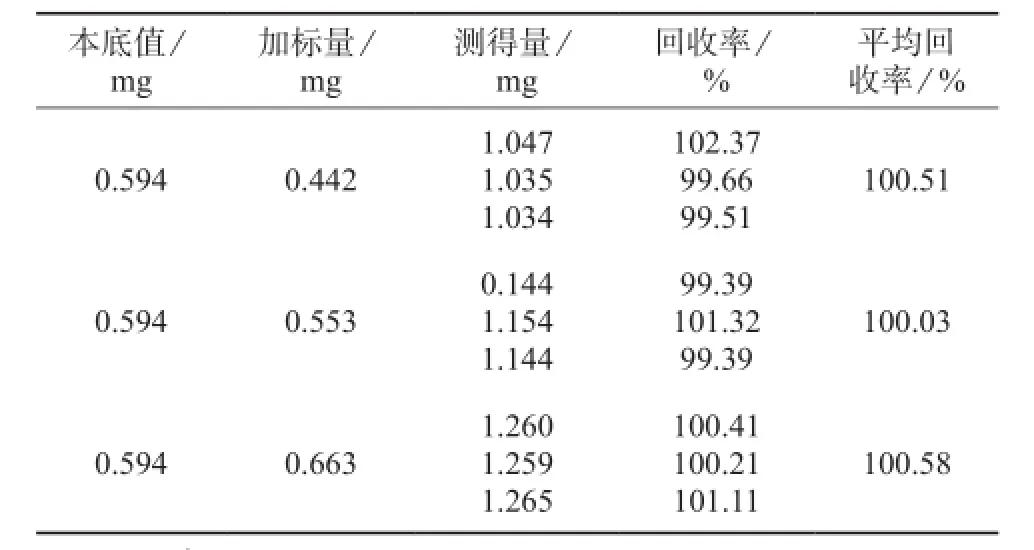
表2 回收试验结果
2.11样品测定
按照优化后核磁共振测试条件,用核磁共振法与药典法(紫外分光光度法)对两个生产厂家的西米替丁片剂样品进行测定,测定结果见表3。由表3可知,本法测定结果与紫外分光光度法测定结果无显著差异,说明所建方法测定结果准确可靠。

表3 核磁共振法与药典法对样品的测定结果(n=5) %
3 结语
采用核磁共振方法测定片剂中西咪替丁含量,该方法的精密度、重复性、稳定性、线性关系及回收率均良好,同时与西咪替丁的药典紫外分光光度法进行了比较,两种方法实验结果相吻合。核磁共振定量法样品用量少,制备过程简单且无需分离,快速准确,是对其它常规定量方法的一种补充,在含量测定的同时可对结构进行确证,对防止假冒伪劣药品的出现发挥一定作用。
[1] Mariusz J S,wiader,Bart omiej Barczyn ski,Micha Tomaszewski,et al. The effects of cimetidine chronic treatment on conventional 4 antiepileptic drugs in mice [J]. Pharmacological Reports,2015,409: 1-6.
[2] Parviz Kokhaei,Mahdieh Shokrollahi Barough,Zuhair M Hassan. Cimetidine effects on the immunosuppression induced by burn injury[J]. International Immunopharmacology,2014,22: 273 -276.
[3] 刘洪海,魏巍,李字.高效液相法测定西咪替丁片的含量[J].药物鉴定,2006,15(12): 15-16.
[4] 方应国,李成平,严小平,等.西咪替丁片剂含量的高效液相色谱法测定[J].药物分析杂志,2010,30(3): 465-467.
[5] Ma Soledad Garcia,Ma Isabel Albero,Concepcion Sa,nchez-Pedren˜o,et al. Spectrophotometric determination of cimetidine in pharmaceuticals and urine using batch and flow-injection methods [J]. Journal of Pharmaceutical and Biomedical Analysis,2003,32: 1 003-1 010.
[6] Diane A I Ashiru,Rajesh Patel,Abdul W Basit. Simple and universal HPLC-UV method to determine cimetidine,ranitidine,famotidine and nizatidine in urine: Application to the analysis of ranitidine and its metabolites in human volunteers [J]. Journal of Chromatography B,2007,860: 235-240.
[7] Volosov A,Alexander C,Ting L,et al.Simple rapid method for quantification of antiretrovirats by liquid chromatography tandem mass-spectrometry.Clin Biochem,2002,35(2): 99-103.
[8] US Pharmacopeia.USP35-NF30[M].USA: United States Pharmacopeia Convention,2012: 2666.
[9] 国家药典委员会.中华人民共和国药典.二部[M].北京:中国医药科技出版社,2015: 348.
[10] 张芬芬,蒋孟虹,沈文斌,等.定量核磁共振(QNMR)技术及其在药学领域的应用进展[J]. 南京师范大学学报: 工程技术版,2014,14(2): 8-18.
[11] 张才煜,吴建敏,李憬,等.核磁共振法定量测定氢溴酸东莨菪碱的绝对含量[J].药物分析杂志,2012,32(2): 327-329.
[12] 杨金霞,姚婷玉,王健伟,等.核磁共振法测定西洛他唑含量[J].化学分析计量,2012,21(4): 68-70.
[13] 张秀丽,王聪,任素梅,等.缓释胶囊中布洛芬含量的核磁共振波谱定量分析方法研究[J].中国海洋药物,2014,33(1): 11-16.
[14] Kenny Kuchta,Jutta Ortwein,Lothar Hennig,et al.1H-qNMR for direct quantification of stachydrine in Leonurus japonicus and L cardiaca[J]. Fitoterapia,2014,96: 8-17.
[15] Chia Yingli,Hong Xixu,Quan Binhan,et al. Quality assessment of Radix Codonopsis by quantitative nuclear magnetic resonance [J]. Journal of Chromatography A,2009,1216: 2 124-2 129.
[16] Siddheshwar K Chauthe,Ram Jee Sharma,Farrukh Aqil,et al. Guptab and Inder Pal Singha Quantitative NMR: An Applicable Method for Quantitative Analysis of Medicinal Plant Extracts and Herbal Products[J]. Phytochem Anal, 2012,23: 689-696.
[17] Malz F,Jancke H. Validation of quantitative NMR[J]. J Pharm Biomed Anal,2005,38: 813-823.
Determination of Cimetidine in Tablets by NMR
Zhang Xiuli, Wang Cong, Wang Yuanhong, Lyu Zhihua
(School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China)
A method for the determination of cimetidine in tables by proton nuclear magnetic resonance spectroscopy (1H NMR) was established. The1H NMR spectra was obtained in MeOD with dimethyl terephthalate as the internal standard by using Agilent DD2-500 MHz spectrometer. For each sample,16 scans were recorded with the following parameters: pulse angle was 45°, relaxation delay was 20 s,acquisition time was 2 s, the temperature was set at 25℃. The linear range was 0.1-5.0 mg/mL with correlation coefficient of 0.999 8. The relative standard deviation of detection results was 0.11%(n=6). The added average recoveries of cimetidine in tables were in the range of 100.03%-100.58%. The cimetidine samples from different factory were detected by the method and pharmacopoeia method, the results were consistent. This method is simple,rapid, with less cimetidine sample, it can be used for the quality control of cimetidine.
cimetidine; nuclear magnetic resonance; tablet; quantitative analysis
O657.2
A
1008-6145(2016)04-0019-04
10.3969/j.issn.1008-6145.2016.04.005
*中央高校基本科研业务费实验室研究基金项目(201651008)
联系人:吕志华;E-mail: lvzhihua@ouc.edu.cn
2016-04-19
猜你喜欢
云南化工(2023年7期)2023-08-01 07:59:34
现代临床医学(2022年4期)2022-09-29 07:36:10
中国抗生素杂志(2022年7期)2022-08-18 03:22:36
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
云南化工(2021年10期)2021-12-21 07:33:42
长春师范大学学报(2019年4期)2019-04-29 05:51:36
家庭医学(2016年3期)2016-04-05 22:23:01
中国卫生标准管理(2015年14期)2016-01-15 02:58:35
河南科技(2015年2期)2015-02-27 14:20:35
同位素(2014年2期)2014-04-16 04:57:13
