
3-芳基-2-硝基丙醇的合成新方法
2016-08-16 07:50李齐激杨小生1
山地农业生物学报 2016年3期
梁 弘,李齐激,杨 艳,潘 雄,杨 娟,杨小生1,*
(1.贵州大学 药学院,贵州 贵阳 550002;2.贵州省中国科学院天然产物化学重点实验室,贵州 贵阳550002)
3-芳基-2-硝基丙醇的合成新方法
梁弘1,2,李齐激2,杨艳2,潘雄2,杨娟2,杨小生1,2*
(1.贵州大学 药学院,贵州 贵阳 550002;2.贵州省中国科学院天然产物化学重点实验室,贵州 贵阳550002)
研究 β-硝基醇类化合物3-芳基-2-硝基丙醇的合成新方法。在碱催化作用下,2-芳基硝基乙烷和甲醛进行缩合,筛选、优化2-芳基硝基乙烷与甲醛反应所需碱的类型和温度,合成3-芳基-2-硝基丙醇。以醋酸钠作为碱,反应温度为30℃时,2-芳基硝基乙烷与过量甲醛能高收率地反应生成3-芳基-2-硝基丙醇。该合成方法有效和反应条件易控,可用于3-芳基-2-硝基丙醇的批量制备。
3-芳基-2-硝基丙醇;甲醛;醋酸钠
β-硝基醇是重要的有机合成中间体,可以进一步转化为许多重要的产物,如还原得到 β-氨基醇、脱水得到硝基的烯烃化合物,氧化得到硝基的羰基化合物,因而被广泛地应用于各类医药中间体和天然产物的合成[1-4]。β-硝基醇的合成方法研究报道较多,主要采用Henry反应[1],不同类型的醛和硝基甲烷,选择不同的手性碱催化剂,可得到不同的手性硝基醇类化合物[5-6]。据文献调研,多数是以硝基甲烷与一定的醛类底物反应,不同类型的硝基烷与甲醛进行Henry反应的报道较少,Fuganti C等[7]利用生物催化对映选择性合成了3-芳基-2-硝基丙醇化合物,且用NaOH作为催化剂以中低收率获得了相应的消旋体化合物,但没有开展合成衍生物研究。在我们的合成研究工作中,3-芳基-2-硝基丙醇为关键中间体。以邻氯硝基苯乙烷[8]为原料,NaOH作为碱,室温下,与甲醛水溶液反应,虽得到了相应的目标产物3-邻氯苯基-2-硝基丙醇,但也产生了较多量的双甲醛加合物。

图1 2-邻氯苯基硝基乙烷与甲醛反应产物
为达到此类反应产物选择性高、反应条件易于控制的效果,我们在试剂甲醛过量的情况下,筛选、优化碱试剂的类型和反应温度条件。
1 材料与方法
1.1试剂及仪器
油浴反应器(上海耀特仪器厂),真空干燥机(天津泰斯特仪器有限公司),INOVA400核磁共振波谱仪(美国瓦里安公司,TMS为内标),HP-5973型质谱仪测定分子量(惠普公司),BuchiR-210型旋转蒸发仪(瑞士布奇公司),磁力搅拌器(上海司乐仪器有限公司)。本实验所以试剂均为天津市科密欧化学试剂有限公司分析纯产品。
1.2合成方法及路线
参照文献方法[8]合成2-芳基硝基乙烷。以甲醇作溶剂,2-芳基硝基乙烷在碱的作用下,与甲醛发生羟醛缩合而得到消旋体3-芳基-2-硝基丙醇及其双甲醛加成产物,合成路线如图2。
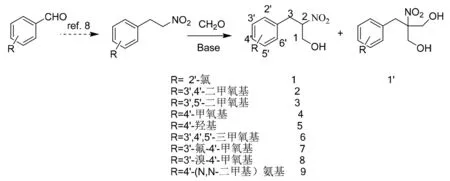
图2 3-芳基-2-硝基丙醇的合成路线
1.3碱的筛选
以2-邻氯苯基硝基乙烷为原料进行碱的类型选择。
称取2-邻氯苯基硝基乙烷50 mg(0.23 mmol )于反应瓶中,氮气保护下,加入甲醛水溶液(0.5 mL,1.3 mmol ) ,甲醇(15mL) ,分别加入不同的碱(0.5 mmol)(碳酸钾,碳酸氢钠,二乙胺,氨水,三乙胺,无水乙酸钾,无水乙酸钠和乙酸铵)搅拌使之完全溶解,继续室温下搅拌4 h,每隔0.5 h以TLC检测反应进度。反应完成后,以乙酸乙酯(100 mL) 萃取2次,合并有机层,乙酸乙酯层依次以水(100 mL×2) 、饱和氯化钠水溶液 (100 mL×2) 洗涤,有机层以无水MgSO4干燥,减压回收乙酸乙酯得粗产物。减压柱层析分离纯化。
1.4温度的选择
称取2-(邻氯苯基)硝基乙烷50 mg( 0.23 mmol)于反应瓶中,氮气保护下,加入甲醛水溶液(0.5 mL,1.3 mmol ) ,甲醇(15 mL ) ,醋酸钠(0.5 mmol)搅拌使之完全溶解,在冰浴0-5℃、油浴30℃、油浴60℃条件下,搅拌反应,每隔0.5 h以TLC检测反应进度。
1.5不同底物的反应
称取不同取代的2-芳基硝基乙烷50 mg (0.23 mmol )于反应瓶中,氮气保护下,加入甲醛水溶液(0.5 mL,1.3 mmol ) ,甲醇(15 mL) ,分别加入醋酸钠(0.5 mmol)搅拌使之完全溶解,于室温拌4 h,每隔0.5 h以TLC检测反应进度。反应完成后,以乙酸乙酯(100 mL) 萃取2次,合并有机层,乙酸乙酯层依次以水(100 mL×2) 、饱和氯化钠水溶液 (100 mL×2) 洗涤,有机层以无水MgSO4干燥,减压回收乙酸乙酯得粗产物。减压柱层析分离纯化。
2 结果与分析
2.1使用不同的碱催化的结果
通过2-(邻氯苯基)硝基乙烷与甲醛缩合采用不同的碱对此反应进行催化,反应主要生成产物及收率见表1。
从表1可以看出以2倍量的醋酸钠作为碱时,无论是产物的单一性,还是收率,均为最佳。
2.2温度的选择结果
通过在不同温度下2-(邻氯苯基)硝基乙烷与甲醛缩合并且使用醋酸钠作为碱催化,对终产物的结构随温度和时间的变化的影响做了考察。结果如表2所示。

表1 不同碱催化所得产物及收率结果
注:a:单一产物;b:柱层析分离后的得率。
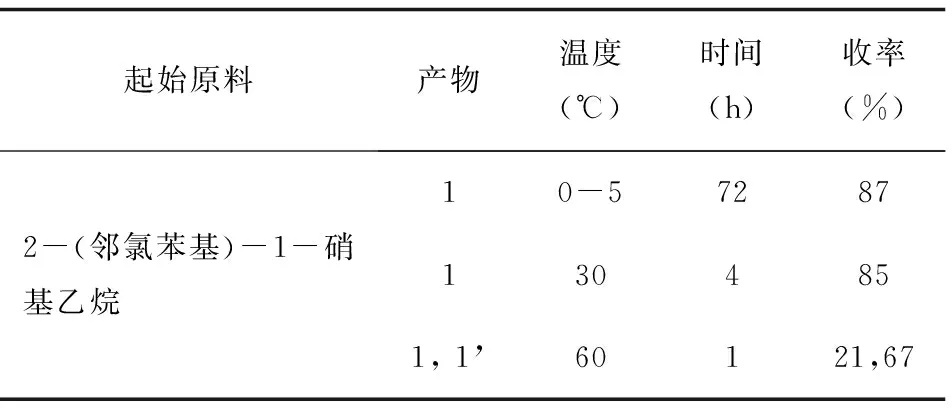
表2 不同温度反应结果
其他条件一致,改变温度情况下,进行反应。表2结果显示,在冰浴(0-5℃)条件时,反应进行缓慢,完全反应需要3 d。在温度为30℃时,4 h反应能进行完全,且产物单一,为化合物1。在温度为60℃时,0.5 h时经TLC检测发现原料还未反应完全,与对照品对照,发现化合物1和化合物1’已生成;1.0 h后,反应完全,但化合物1’已成为主要产物。
2.3不同底物的反应结果
使用不同取代的2-芳基硝基乙烷与甲醛在醋酸钠催化下进行缩合,各硝基乙烷反应完成后的结果如表3所示。
从表中可看出,在醋酸钠为碱,室温下,相应的硝基烷与过量甲醛水溶液反应,可高收率得到3-芳基-2-硝基丙醇类化合物。

表3 不同原料得到的产物及收率
2.4各化合物的合成及波谱数据
化合物1 的合成及其波普数据:
称取2-邻氯苯基硝基乙烷50 mg(0.23 mmol )于反应瓶中,氮气保护下,加入甲醛水溶液(0.5 mL,1.3 mmol ) ,甲醇(15 mL) ,分别加入醋酸钠(0.5 mmol)(碳酸钾,碳酸氢钠,二乙胺,氨水,三乙胺,无水乙酸钾,无水乙酸钠铵)搅拌使之完全溶解,继续室温下搅拌4 h,每隔0.5 h以TLC检测反应进度。反应完成后,以乙酸乙酯(100 mL) 萃取2次,合并有机层,乙酸乙酯层依次以水(100 mL×2) 、饱和氯化钠水溶液 (100 mL×2) 洗涤,有机层以无水MgSO4干燥,减压回收乙酸乙酯得粗产物。减压柱层析分离纯化。得到化合物1即3-邻氯苯基-2-硝基丙醇收率85%。
ESI - MSm/z: 238 [M+Na]+;1H NMR (400MHz,CDCl3) δ: 7.51 - 7.15 (m,4H,-Ar),5.02-4.88 (m,1H,-CHNO2),4.05-3.97 (m,2H,-CH2OH),3.44-3.22 (m,2H,-CH2Ar);13C NMR (100MHz,CDCl3) δ: 134.0 (C-1’) ,132.6 (C 2’) ,131.4 (C-3’) ,129.8 (C-6’),129.2 (C-4’) ,127.3 (C-5’) ,87.9 (C-2),62.5 (C-1),33.6 (C-3) .
化合物2 的合成及其波普数据:
化合物2依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以86%的收率得到化合物2即3-(3’,4’-二甲氧基苯基)-2-硝基丙醇。
ESI - MSm/z: 264[M+Na]+;1H NMR (400MHz,CD3COCD3) δ:7.01 - 6.73 (m,3H,-Ar),5.01 - 4.84 (m,1H,-CHNO2),4.04 - 3.92 (m,2H,-CH2OH),3.79 (s,3H,-OCH3),3.77 (s,3H,-OCH3),3.13 - 3.07 (m,2H,-CH2Ar);13C NMR (100 MHz,CD3COCD3) δ :150.3 (C-3’),149.4 (C-4’),129.1 (C-1’),121.8 (C-6’),113.5 (C-2’),112.6 (C-5’),92.2 (C-2),63.5 (C-1),55.9 (-OCH3),35.9 (C-3).
化合物3 的合成及其波普数据:
化合物3 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以87%的收率得到化合物3即3-(3’,5’-二甲氧基苯基)-2-硝基丙醇
ESI - MSm/z: 264 [M+Na]+;1H NMR (400 MHz,CD3OD) δ : 6.34 (s,3H,-Ar),4.89 - 4.81 (m,1H,-CHNO2),3.89 (ddd,J= 15.8,12.1,6.0 Hz,2H,-CH2OH),3.71 (s,6H,-OCH3×2),3.02 (ddd,J= 19.9,14.2,7.4 Hz,2H,-CH2Ar);13C NMR (100 MHz,CD3OD) δ : 162.4 (C-3’,C-5’) ,139.2 (C-1’) ,107.8 (C-2’,C-6’) ,99.9 (C-4’) ,92.2 (C-2) ,63.7 (C-1) ,55.6 (-OCH3),36.8 (C-3) .
化合物4 的合成及其波普数据:
化合物4 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以87%的收率得到化合物4即3-(4’-甲氧基苯基)-2-硝基丙醇。
ESI - MSm/z: 234[M+Na]+;1H NMR (400MHz,CD3COCD3) δ : 7.27 - 6.78 (m,4H,-Ar),4.97 - 4.79 (m,1H-CHNO2),4.12 - 3.86 (m,2H,-CH2OH),3.76 (s,3H,-OCH3),3.12 - 3.07 (m,2H-CH2Ar).13C NMR (100 MHz,CD3COCD3) δ: 159.7 (C-4’) ,130.7 (C-1’) ,128.5 (C-2’,C-6’),114.7 (C-3’,C-5’) ,92.3 (C-2) ,63.6 (C-1) ,55.3 (-OCH3) ,35.4 (C-3) .
化合物5 的合成及其波普数据:
化合物5 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以83%的收率得到化合物5即3-(4’-羟基苯基)-2-硝基丙醇。
ESI - MSm/z: 220[M+Na]+;1H NMR (400 MHz,CD3COCD3) δ 7.14 - 6.72 (m,4H,-Ar),4.93 - 4.76 (m,1H-CHNO2),4.07 - 3.85 (m,2H,-CH2OH),3.13 - 3.03 (m,2H,-CH2Ar);13C NMR (100 MHz,CD3COCD3) δ :157.3 (C-4’),130.8 (C-1’),127.3 (C-2’,C-6’),116.2 (C-3’,C-5’),92.4 (C-2),63.6 (C-1),35.5 (C-3).
化合物6的合成及其波普数据:
化合物6 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以84%的收率得到化合物6即3-(3’,4’,5’-三甲氧基苯基)-2-硝基丙醇。
ESI - MSm/z: 294[M+Na]+;1H NMR (400MHz,CD3OD) δ : 6.50 (s,2H,-Ar),4.93 - 4.85 (m,1H,-CHNO2),3.97 - 3.85 (m,2H,-CH2OH),3.77 (s,6H,-OCH3×2),3.71 (s,3H,-OCH3),3.13 - 3.00 (m,2H,-CH2Ar);13C NMR (100MHz,CD3OD)δ:154.5 (C-3’,5’),138.0 (C-4’),133.1 (C-1’),107.1 (C-2’,C-6’),92.3 (C-2),63.7 (C-4’-OCH3),61.0[ (C-3’,C-5’)-OCH3],56.5 (C-1),36.8 (C-3).
化合物7的合成及其波普数据:
化合物7 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以86%的收率得到化合物6即 3-(3’-氟-4’-甲氧基苯基)-2-硝基丙醇
ESI - MS ,m/z: 252[M+Na]+;1H NMR (400MHz,CD3OD) δ :7.10 - 6.81 (m,3H,-Ar),4.84 - 4.78 (m,1H,-CHNO2),3.95 - 3.84 (m,2H,-CH2OH),3.83 (s,3H,-OCH3),3.17 - 2.91 (m,2H,-CH2Ar).13C NMR (400MHz,CD3OD) δ : 154.4 (C-3’),152.5 (C-4’),129.9 (C-1’),126.0 (C-6’),117.4 (C-2’),114.8 (C-5’),92.3 (C-2),63.7 (-OCH3),56.6 (C-1),35.6 (C-3).
化合物8的合成及其波普数据:
化合物8 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以90%的收率得到化合物6即 3-(3’-溴-4’-甲氧基苯基)-2-硝基丙醇
ESI - MS ,m/z: 311.99[M+Na]+;1H NMR (400MHz,CD3OD) δ : 7.39 (s,1H,-Ar),7.19 - 7.10 (m,1H,-Ar),6.95 - 6.88 (m,1H,-Ar),4.85 - 4.71 (m,1H-CHNO2),4.03 - 3.87 (m,2H,-CH2OH),3.84 - 3.76 (m,3H,-OCH3),3.14 - 2.86 (m,2H,-CH2Ar);13C NMR (400 MHz,CD3OD) δ:156.5 (C-4’),134.5 (C-1’),130.5 (C-2’),130.3 (C-6’),113.2 (C-5’),112.4 (C-3’),92.3 (C-2),63.68(-OCH3),56.6 (C-1),35.3 (C-3).
化合物9的合成及其波普数据:
化合物9 依据上述化合物1即3-邻氯苯基-2-硝基丙醇的合成方法以81%的收率得到化合物6即3-(4’-二甲氨基苯基)-2-硝基丙醇.ESI - MS ,m/z:247[M+Na]+;1H NMR (400 MHz,CD3OD) δ: 7.27 - 6.87 (m,4H,-Ar),4.83 - 4.75 (m,1H,-CHNO2),3.94 - 3.84 (m,2H,-CH2OH),3.83 (s,3H,-OCH3),3.81 (s,3H,-OCH3),3.08 - 2.95 (m,2H,-CH2Ar);13C NMR (400 MHz,CD3OD) δ: 156.4 (C-4’),136.3 (C-1’),134.5 (C-2’),133.0 (C-5’),130.2 (C-3’),128.5 (C-6’),92.2 (C-2),64.0(-NCH3),63.6(-NCH3),56.6 (C-1),35.3 (C-3).
化合物1’的合成及其波普数据:
称取2-邻氯苯基硝基乙烷50 mg(0.23 mmol)于反应瓶中,氮气保护下,加入甲醛水溶液(0.5 mL,1.3 mmol) ,甲醇(15 mL) ,分别加入不同的碱(0.5 mmol)碳酸氢钠,搅拌使之完全溶解,继续室温下搅拌4 h,每隔0.5 h以TLC检测反应进度。反应完成后,以乙酸乙酯(100 mL) 萃取2次,合并有机层,乙酸乙酯层依次以水(100 mL×2) 、饱和氯化钠水溶液 (100 mL×2) 洗涤,有机层以无水MgSO4干燥,减压回收乙酸乙酯得粗产物。减压柱层析分离纯化。得到化合物1’即3-邻氯苯基-2-硝基-2-羟甲基丙醇收率85%。
ESI - MS ,m/z:268[M+Na]+1 ;1H NMR (400 MHz,CD3COCD3) δ7.48 - 7.27 (m,4H,-Ar),4.11 - 3.97 (m,4H-CH2OH×2),3.50 (s,2H,-CH2Ar).13C NMR (400MHz,CD3COCD3) δ: 135.5 (C-1’),133.6 (C-2’),133.2 (C-3’),130.3 (C-6’),129.8 (C-4’),127.8 (C-5’),95.5 (C-2),62.1 (C-1,C-1’),34.4 (C-3).
3 结论与讨论
本实验的目的在于找到合适的合成芳香硝基醇的方法,进而合成一系列的芳基硝基醇,且作为还原合成氨基醇的中间体。与其他使用氨基酸还原合成氨基醇的方法路线相比[ 5 ],此路线更优越的地方在于:
(1)能实现芳基上不同位置取代的多样性,不受氨基酸结构的局限。能定向的对芳香氨基醇的芳环进行修饰;
(2)不需使用LiAlH4等较易然的试剂;
(3)直接还原产率受氨基酸溶解度等的影响,部分产率极低,有些优化只能达到49.93%[6]。
(4)本文合成的产物均为外消旋产物,但有文献报道直接可以利用生物酶催化技术将外消旋体分开[7]。
虽有文献[7]报道采用硝基烷通过碱催化并与甲醛反应合成硝基醇,但随取代基的不同其收率变化幅度较大,且收率均不高,为48-71%。而本实验的收率均在80%以上,达到了优化收率的目的。并且为从事后期的合成工作找到的最适宜的路线。
通过设计、合成了系列3-芳基-2-硝基丙醇类化合物。在醋酸钠为碱,室温下,相应的硝基烷与过量甲醛水溶液反应,可高收率得到3-芳基-2-硝基丙醇类化合物。与文献[1,7]相比,反应条件温和、合成产物选择性和收率高,操作条件易控且易于大量制备。虽然本实验终产物为外消旋体,但现可用生物酶催化技术将其快速分开[7]。本实验结果为中间体原料硝基醇及氨基醇的合成都提供了可靠选择方法。
致谢:感谢贵州省中国科学院天然产物化学重点实验室分析测试中心提供的波普数据。
[1] Luzzio F. A. The Henry reaction: recent examples [J].Tetrahedron,2001,57(6): 915-945.
[2]叶秀林. 立体化学[M]. 北京: 北京大学出版社,1997: 65.
[3]张坤,晏菊芳,唐雪梅,等. 含萘丁美酮结构单元的 β-氨基醇的合成及其抗糖尿病活性研究[J]. 药学学报,2011,46(4): 412-421.
[4]司曼. 去甲肾上腺素和血管紧张素 Ⅱ 对心肌慢激活延迟整流钾电流的调节作用及机制[D]. 石家庄:河北医科大学,2014.
[5]李高伟,赵文献,郭蕊,等. 基于烷基锌催化的不对称Henry反应研究进展[J].化学研究, 2010,21(5): 107-112.
[6]甘昌胜,潘见. 不对称催化Henry反应研究进展[J].有机化学, 2008,28(7): 1193-1198.
[7] Fuganti C,Sacchetti A. Biocatalytic enantioselective approach to 3-aryl-2-nitropropanols: Synthesis of enantioenriched (R)-5-methoxy-3-aminochroman,a key precursor to the antidepressant drug Robalzotan[J].JournalofMolecularCatalysisB:Enzymatic,2010,66(3): 276-284.
[8]杨小生,郝小江,周俊. dl-反式-4-苯基-5-邻氯苄基吡咯烷酮-2的合成工艺:CN 97123419.1[P]. 1998-8-26.
A new Method for Synthesis of 3-aryl-2-nitropropanol
LIANGHong1,2,LIQi-ji2,YANGYan2,PANXiong2,YANGJuan2,YANGXiao-sheng2*
(1.CollegeofPharmacy,GuizhouUniversity,Guiyang,Guizhou550025,China; 2.TheKeyLabofChemistryforNaturalProductsofGuizhouProvinceandChineseAcademyofSciences,Guiyang,Guizhou550002,China)
Here we reported a new synthetic method for 3-aryl-2-nitropropanols. Different bases and temperature condition were screened and optimized for the condensation reaction of 2-aryl nitroethane with formaldehyde in methanol. (±) 3-aryl-2-nitropropanols were obtained in high yields by reaction conditions with sodium acetate as base and temperature at 30℃ . The synthetic method was effective and available for a large amount of production of 3-aryl-2-nitropropanol.
3-Aryl-2-nitropropanol; Formaldehyde; Sodium acetate
1008-0457(2016)03-0044-05国际DOI编码:10.15958/j.cnki.sdnyswxb.2016.03.008
2016-03-22;修回日期:2016-04-12
贵州省高层次创新型人才培养(百层次)(黔科合人才[2015]4027号)。
杨小生(1966-),男,博士,研究员,博士生导师,主要研究方向:重要天然活性物质研究和药物合成,中药民族药中活性物质研究与开发; E-mail: gzcnp@sina.cn。
Q621.3
A
猜你喜欢
分子催化(2022年1期)2022-11-02
食品工业科技(2021年23期)2021-12-16
煤气与热力(2021年3期)2021-06-09
石油化工建设(2018年2期)2018-07-11
铜仁学院学报(2018年6期)2018-07-05
食品与生活(2017年5期)2017-05-27
合成化学(2015年2期)2016-01-17
合成化学(2015年10期)2016-01-17
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
